Types
There are a large number of neurocutaneous syndromes that exceed the scope of this article. Therefore, characteristics of a few of the more common types are summarized.
Neurofibromatosis Type I (von Recklinghausen disease)


Neurofibromatosis type 1 is the most common phakomatosis and it affects approximately 1 in 2500-3000 live births. [9] It is a genetic disorder due to a germline mutation in the NF1 gene. This gene encodes a protein called neurofibromin that is involved in controlling cellular growth. [10] Malfunction of the gene results in multisystem manifestations involving the skin, central nervous system, peripheral nervous system, eyes and musculoskeletal system. The condition is inherited in an autosomal dominant manner. However, approximately one-half of patients with this condition have no family history and the mutation occurs spontaneously. In most instances, neurofibromatosis type 1 can be diagnosed clinically according to consensus criteria and genetic testing is only used in atypical presentations or for family planning decisions. [11]
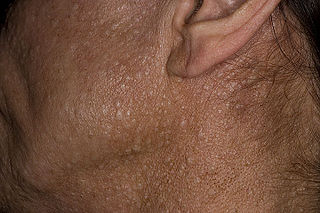
Café au lait spots are one of the most characteristic features of neurofibromatosis type 1. They are hyperpigmented lesions of the skin that increase in number and size during the first years of life. They are present in almost all patients but do not have malignant potential. [12] [13] Freckling in the axillary and inguinal regions are another common presenting feature seen in as many as 90% of patients during childhood. [12] Lisch nodules (benign hamartomas of the iris) are seen in almost all patients but they do not cause any visual or ocular impairment.
Neurofibromas are benign nerve sheath tumors that occur in peripheral nerves. These typically develop during the teenage years. Neurofibromas do not become malignant but can cause cosmetic concerns as well as local pruritus. When present in the spine they can affect nerve roots and result in both motor and sensory defects. [13] Surgery may be required in some cases. On the contrary, plexiform neurofibromas arise from multiple nerve fascicles and malignant transformation occurs in approximately 10% of cases. [14] They may result in significant morbidity as they may cause organ compression, vascular occlusions, bone destruction, pain and cosmetic issues. Plexiform neurofibromas are seen in 30-50% of patients. [12]
Optic pathway gliomas are seen in 15-20% of patients with neurofibromatosis type 1. [12] They most often arise during childhood. They often do not cause additional morbidity but may in up to one-half of patients. [15] Neurocognitive impairment, attention-deficit hyperactivity disorder, autism spectrum disorder and behavioral disorders are also commonly seen in patients with neurofibromatosis type 1. [16] Epilepsy is seen in 4-7% of patients. [17]
Musculoskeletal system manifestations can develop in patients with neurofibromatosis type 1. Common findings include sphenoid wing dysplasia, osteopenia, osteoporosis, anterior chest wall deformities as well as scoliosis. Patients are at a much greater risk for fractures than the general population. [13]
Vascular abnormalities are also frequently encountered in patients with neurofibromatosis type 1. These may include renal artery stenosis, pulmonary artery stenosis, cerebral artery stenosis and aneurysms. [13] Complications may include myocardial infarction and stroke. [18]
Relative to the general population the risk for certain types of cancer is increased significantly. For instance, the risk for brain tumors and breast cancer is increased by 5 times. [13]
Neurofibromatosis Type II

Neurofibromatosis type 2 is an autosomal dominant condition that affects approximately 1 in 35,000-40,000 people. [20] It is caused by mutations in the NF2 gene on chromosome 22 which has a high penetrance though most patients do not present with symptoms until adulthood. [21] Approximately half of patients have de novo mutations and as many as 59.7% are mosaic. [22] [23] Patients who present in childhood tend to have a more severe phenotype. [24]
Bilateral vestibular schwannomas are the most characteristic finding and in the vast majority of cases are benign. However, they are responsible for significant morbidity and are responsible for the most common presenting symptom of hearing loss in adults. [25] They can also less commonly present with dizziness or balance impairment. It is estimated that schwannomas occur in over 90% of patients. [26]
Meningiomas are the second most common tumor in NF2. Approximately half of patients have an intracranial meningioma and extramedullary spinal meningiomas occur in one-fifth of patients. [25] Relative to the general population, meningiomas in NF2 tend to occur at a younger age, are more likely to be multiple and are associated with increased mortality. [27]
Spinal cord ependymomas occur in 20-50% of patients but they are asymptomatic in the majority of cases. [25] In cases that they are symptomatic the specific presentation will depend upon where they are located. Potential symptoms include back pain, weakness and sensory changes.
Peripheral neuropathy eventually occurs in most patients with NF2. In some cases, a mononeuropathy affecting the facial nerve can occur as the first presenting symptom of NF2. [25] Other potential manifestations include focal amyotrophy, mononeuropathy multiplex or a severe generalized polyneuropathy in 3-5% of patients. [22]
Ophthalmologic manifestations are also common with 60-80% of patients developing cataracts. [25] Optic nerve meningiomas and retinal hamartomas can result in vision loss.
Approximately, 70% of patients will have cutaneous manifestations but only 10% have more than 10 lesions. [25] The majority of skin lesions are schwannomas but neurofibromas can also occur.
Tuberous sclerosis (Bourneville syndrome)


Tuberous sclerosis complex (TSC) is a multisystemic disorder due to autosomal dominant mutations in either TSC1 or TSC2 which results in the impaired inhibition of the mechanistic target of rapamycin (mTOR) signaling pathway. [28] This leads to impaired regulation of cellular proliferation, survival, homeostasis, migration and other critical functions. [29] The most typically affected organs include the brain, skin, kidney, heart and lung. The incidence of TSC is approximately 1 in 6,000 live births. [30] Similar to other neurocutaneous disorders there is variable penetrance and expressivity. [28] TSC1 mutations tend to have a less severe phenotype and are more likely to be familial. [28] A major development in the treatment of this condition occurred in 2010s when the FDA approved mTOR inhibitors for the treatment of several manifestations of TSC.
Epilepsy is among the most common manifestations of TSC and it occurs 80-90% of patients. [31] It usually presents during the first few years of life and is medically refractory in two-thirds of patients. [32] Approximately, one-third of patients have infantile spasms. [32] There are several types of brain lesions that can be found in TSC including subependymal nodules (SENs), cortical tubers and subependymal giant cell astrocytomas (SEGAs). SENs and cortical tubers occur in approximately 80% and 90% of patients, respectively. [33] SEGAs occur in 10-15% of patients and are a major potential cause of morbidity and potentially mortality. [34] They tend to present during the first 20 years of life. TSC-Associated Neuropsychiatric Disorder (TAND) refers to the behavioral, intellectual and psychiatric manifestations of TSC including autism spectrum disorder, attention deficit hyperactivity disorder, intellectual disability, depression and anxiety. Approximately 90% of patients will have at least 1 symptom of TAND. [35]
Lymphangioleiomyomatosis (LAM) occurs in the lung and may result in pneumothorax, cystic lung destruction and pleural effusions. Symptoms which occur as a result may include fatigue, chest pain and shortness of breath. It occurs in approximately 30-40% of women with TSC, up to 80% by the age of 40, and it is much less common in men. [36]
Renal angiomyolipomas and cysts are the most common manifestations of TSC involving the kidney. Renal disease is among the most common causes of early death in TSC. One study found that renal lesions were present in 80% of patients by a mean age of 10.5 years. [37] Renal cell carcinoma occurs in 2-5% of patients with TSC at a mean age of 28–30 years. [38]
Cardiac rhabdomyomas are benign hamartomas and are the most common cardiac manifestations of TSC. It is found in approximately two-third of newborns with TSC. [39] Most of the time they do not cause symptoms and spontaneously regress. In a minority of cases, they may result in heart failure, arrhythmias, and murmurs. [39]
Dermatological manifestations occur in almost all patients and may include facial angiofibromas, confetti skin lesions, ungual fibromas, shagreen patches, hypomelanotic macules and fibrous cephalic plaques.[13] None of these tend to result in significant complications however facial angiofibromas may cause significant cosmetic concerns. [40]
Sturge–Weber Syndrome

Sturge-Weber syndrome occurs in approximately 1 in 20,000-50,000 live births and is caused by a somatic activating mutation in GNAQ. [41] [42] Normally, GNAQ is involved in cell growth signal transmission. [42] It is classically characterized by a facial port-wine stain in the ophthalmic division of the trigeminal nerve, glaucoma and leptomeningeal angioma. [41] However, the clinical presentation can vary significantly depending on the timing of the somatic mutation ranging from an isolated port-wine stain to complete Sturge-Weber syndrome. [41]
The characteristic port-wine stain, also called nevus flammeus, is caused by a capillary or venular malformation. It is present from birth while the extent and size of it is associated with the risk of leptomeningeal and ophthalmologic involvement. [43] The greatest risk is associated with port-wine stains that appear to involve the entire V1 distribution followed by partial V1 involvement. [43] There is ontroversy as to whether or not the distribution of port-wine stains truly follows trigeminal nerve branches per se. [44] Port-wine stains are most often unilateral but can be bilateral. Typically, brain and eye involvement occur on the same side as the port-wine stain. Over time, port-wine stains can develop soft tissue or bone hypertrophy, proliferative nodules, and progressive ectasia which can lead to significant disfigurement. [45]
The two most common ocular manifestations include glaucoma and choroidal hemagioma. Glaucoma is estimated to occur in 30-70% with a bimodal peak occurring at the time of birth in 60% and between childhood and adolescence in 40%. [46] The most frequent form of glaucoma is open-angle however closed-angle glaucoma may also occur. [46] Congenital onset glaucoma is associated with other changes of the eye including megalocornea and buphthalmos. [47] The choroidal hemangioma occurs in 40-50% of patients. [48] They are usually asymptomatic but they may become thickened over time and may be associated with an increased risk for glaucoma. [46] [49]
Seizures typically develop within the first two years of life and occur in 75-100% of patients. [50] [41] Onset of seizures occur in 95% by the age of 5 years. [51] Status epilepticus occurs in approximately 50% of patients. [52] Approximately 25% of patients develop drug-resistant epilepsy. [53] In these cases, surgery may be pursued with the two main approaches being lesionectomy and hemispherectomy. [53]
Neurological impairment can accrue gradually over time and may occur in the context of stroke-like episodes which may be triggered by seizures or head injuries. [50] Intellectual disability occurs in approximately one-half of patients. [50] In addition, patients with Sturge-Weber syndrome have an increased prevalence of depression, endocrinological abnormalities, headaches with migraine-like features, ADHD and behavioral problems. [54] Cortically mediated visual field defects and hemiparesis occur in approximately one-third and one-half of patients, respectively. [50]
Von Hippel–Lindau syndrome (hemangiomatosis)

Von Hippel-Lindau (VHL) disease is an autosomal dominant condition caused by mutations of the VHL gene. [56] Approximately one-in-five cases are de novo rather than familial and it has nearly complete penetrance. [57] VHL occurs in an estimated 1 in 36,000-45,000 live births. [57] VHL normally functions as a tumor suppressor gene and thus when not functioning normally results in the development of benign and malignant tumors as well as cysts. Some of the most common manifestations include hemangioblastomas in the retina and central nervous system, clear cell renal cell carcinomas, pheochromocytomas, endolymphatic sac tumors and pancreatic neuroendocrine tumors. Life expectancy is reduced for individuals with this condition and one study found a median age of death at 52 years. [58]
CNS hemangioblastomas are the cardinal feature of VHL and occur in as many as 80% of cases. [59] The most common locations include the cerebellum, spinal cord and brainstem. [59] These tumors are benign however they may cause significant morbidity as well as mortality due to mass effect. [60] Tumor growth rates can be highly variable. Treatment options include resection and radiation. [60]
Retinal hemangioblastomas occur in approximately half of patients and are often the first presenting manifestation of VHL. [61] One longitudinal study that followed patients for a mean period of 7.3 years found that in individuals with unilateral disease 100% had bilateral involvement by the age of 56 years. [62] Blindness or severe visual impairment occurs in less than 10% of patients. [63] [64] Laser photocoagulation and cryotherapy are the most common surgical treatments. [59]
Renal cell carcinoma is a major cause of death in patients with VHL and it occurs in 70% of patients. [59] Lesions larger than 3 cm are associated with a risk of metastasis and thus present a recommended threshold for resection. [65] [66] Nephron sparing surgery allows for preservation of kidney function and 10-year survival rates up to 81%. [67]
Additional manifestations of VHL may include pheochromocytomas in as many as 16% of patients with VHL. [68] They are most often unilateral but can be bilateral or multifocal. Approximately 5% are malignant. [59]
Pancreatic involvement occurs in 77% of patients with VHL. Asymptomatic cysts consist of the majority of cases. Neuroendocrine tumors occur in approximately 15% of cases. [69] Less than 10% with neuroendocrine tumors will develop metastases. [70]














{kind=link}