A brain tumor occurs when abnormal cells form within the brain. There are two main types of tumors: malignant tumors and benign (non-cancerous) tumors. These can be further classified as primary tumors, which start within the brain, and secondary tumors, which most commonly have spread from tumors located outside the brain, known as brain metastasis tumors. All types of brain tumors may produce symptoms that vary depending on the size of the tumor and the part of the brain that is involved. Where symptoms exist, they may include headaches, seizures, problems with vision, vomiting and mental changes. Other symptoms may include difficulty walking, speaking, with sensations, or unconsciousness.

A teratoma is a tumor made up of several different types of tissue, such as hair, muscle, teeth, or bone. Teratomata typically form in the tailbone, ovary, or testicle.
Spinal tumors are neoplasms located in either the vertebral column or the spinal cord. There are three main types of spinal tumors classified based on their location: extradural and intradural. Extradural tumors are located outside the dura mater lining and are most commonly metastatic. Intradural tumors are located inside the dura mater lining and are further subdivided into intramedullary and extramedullary tumors. Intradural-intramedullary tumors are located within the dura and spinal cord parenchyma, while intradural-extramedullary tumors are located within the dura but outside the spinal cord parenchyma. The most common presenting symptom of spinal tumors is nocturnal back pain. Other common symptoms include muscle weakness, sensory loss, and difficulty walking. Loss of bowel and bladder control may occur during the later stages of the disease.

Meningioma, also known as meningeal tumor, is typically a slow-growing tumor that forms from the meninges, the membranous layers surrounding the brain and spinal cord. Symptoms depend on the location and occur as a result of the tumor pressing on nearby tissue. Many cases never produce symptoms. Occasionally seizures, dementia, trouble talking, vision problems, one sided weakness, or loss of bladder control may occur.

Germ cell tumor (GCT) is a neoplasm derived from the primordial germ cells. Germ-cell tumors can be cancerous or benign. Germ cells normally occur inside the gonads. GCTs that originate outside the gonads may be birth defects resulting from errors during development of the embryo.
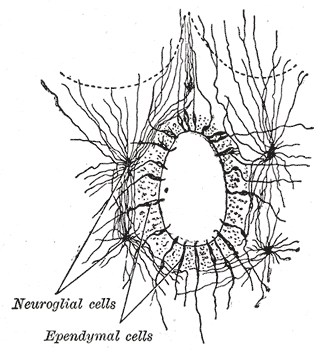
The ependyma is the thin neuroepithelial lining of the ventricular system of the brain and the central canal of the spinal cord. The ependyma is one of the four types of neuroglia in the central nervous system (CNS). It is involved in the production of cerebrospinal fluid (CSF), and is shown to serve as a reservoir for neuroregeneration.

The central canal is the cerebrospinal fluid-filled space that runs through the spinal cord. The central canal lies below and is connected to the ventricular system of the brain, from which it receives cerebrospinal fluid, and shares the same ependymal lining. The central canal helps to transport nutrients to the spinal cord as well as protect it by cushioning the impact of a force when the spine is affected.
Sacrococcygeal teratoma (SCT) is a type of tumor known as a teratoma that develops at the base of the coccyx (tailbone) and is thought to be primarily derived from remnants of the primitive streak. Sacrococcygeal teratomas are benign 75% of the time, malignant 12% of the time, and the remainder are considered "immature teratomas" that share benign and malignant features. Benign sacrococcygeal teratomas are more likely to develop in younger children who are less than 5 months old, and older children are more likely to develop malignant sacrococcygeal teratomas.

Ganglioglioma is a rare, slow-growing primary central nervous system (CNS) tumor which most frequently occurs in the temporal lobes of children and young adults

Spinal cord compression is a form of myelopathy in which the spinal cord is compressed. Causes can be bone fragments from a vertebral fracture, a tumor, abscess, ruptured intervertebral disc or other lesion.
A germinoma is a type of germ-cell tumor, which is not differentiated upon examination. It may be benign or malignant.
Glial tumor is a general term for numerous tumors of the central nervous system, including astrocytomas, ependymal tumors, Oligodendroglioma, and primitive neuroectodermal tumors. The World Health Organization (WHO) classifies tumors into different categories according to severity and recurrence.The first tumor classified as grade I is called pilocytic astrocytoma and it is most commonly observed in children rather than adults. The next tumor is never common in the Dns called diffuse astrocytoma and it is considered a grade II, they are benign, or noncancerous, but can become malignant, meaning cancerous, as the tumor progresses. Grades III and grade IV are considered malignant astrocytomas. Anaplastic astrocytomas are considered by the WHO to be a grade III astrocytoma and Glioblastoma is a grade IV both are referred to high-grade glial tumors.

Leptomeningeal cancer is a rare complication of cancer in which the disease spreads from the original tumor site to the meninges surrounding the brain and spinal cord. This leads to an inflammatory response, hence the alternative names neoplastic meningitis (NM), malignant meningitis, or carcinomatous meningitis. The term leptomeningeal describes the thin meninges, the arachnoid and the pia mater, between which the cerebrospinal fluid is located. The disorder was originally reported by Eberth in 1870. It is also known as leptomeningeal carcinomatosis, leptomeningeal disease (LMD), leptomeningeal metastasis, meningeal metastasis and meningeal carcinomatosis.

The WHOclassification of tumours of the central nervous system is a World Health Organization Blue Book that defines, describes and classifies tumours of the central nervous system (CNS).
Neuro-oncology is the study of brain and spinal cord neoplasms, many of which are very dangerous and life-threatening. Among the malignant brain cancers, gliomas of the brainstem and pons, glioblastoma multiforme, and high-grade astrocytoma/oligodendroglioma are among the worst. In these cases, untreated survival usually amounts to only a few months, and survival with current radiation and chemotherapy treatments may extend that time from around a year to a year and a half, possibly two or more, depending on the patient's condition, immune function, treatments used, and the specific type of malignant brain neoplasm. Surgery may in some cases be curative, but, as a general rule, malignant brain cancers tend to regenerate and emerge from remission easily, especially highly malignant cases. In such cases, the goal is to excise as much of the mass and as much of the tumor margin as possible without endangering vital functions or other important cognitive abilities. The Journal of Neuro-Oncology is the longest continuously published journal in the field and serves as a leading reference to those practicing in the area of neuro-oncology.
Pediatric ependymomas are similar in nature to the adult form of ependymoma in that they are thought to arise from radial glial cells lining the ventricular system. However, they differ from adult ependymomas in which genes and chromosomes are most often affected, the region of the brain they are most frequently found in, and the prognosis of the patients. Children with certain hereditary diseases, such as neurofibromatosis type II (NF2), have been found to be more frequently afflicted with this class of tumors, but a firm genetic link remains to be established. Symptoms associated with the development of pediatric ependymomas are varied, much like symptoms for a number of other pediatric brain tumors including vomiting, headache, irritability, lethargy, and changes in gait. Although younger children and children with invasive tumor types generally experience less favorable outcomes, total removal of the tumors is the most conspicuous prognostic factor for both survival and relapse.

Astroblastoma is a rare glial tumor derived from the astroblast, a type of cell that closely resembles spongioblastoma and astrocytes. Astroblastoma cells are most likely found in the supratentorial region of the brain that houses the cerebrum, an area responsible for all voluntary movements in the body. It also occurs significantly in the frontal lobe, parietal lobe, and temporal lobe, areas where movement, language creation, memory perception, and environmental surroundings are expressed. These tumors can be present in major brain areas not associated with the main cerebral hemispheres, including the cerebellum, optic nerve, cauda equina, hypothalamus, and brain stem.

A central nervous system primitive neuroectodermal tumor, often abbreviated as PNET, supratentorial PNET, or CNS-PNET, is one of the 3 types of embryonal central nervous system tumors. It is considered an embryonal tumor because it arises from cells partially differentiated or still undifferentiated from birth. Those cells are usually neuroepithelial cells, stem cells destined to turn into glia or neurons. It can occur anywhere within the spinal cord and cerebrum and can have multiple sites of origins, with a high probability of metastasis through cerebrospinal fluid (CSF).

Anaplastic oligodendroglioma is a neuroepithelial tumor which is believed to originate from oligodendrocytes, a cell type of the glia. In the World Health Organization (WHO) classification of brain tumors, anaplastic oligodendrogliomas are classified as grade III. In the course of the disease, it can degenerate into highly malignant oligodendroglioma, grade IV. The vast majority of oligodendrogliomas occur sporadically, without a confirmed cause and without inheritance within a family.
Embryonal tumor with multilayered rosettes (ETMR) is an embryonal central nervous system tumor. It is considered an embryonal tumor because it arises from cells partially differentiated or still undifferentiated from birth, usually neuroepithelial cells, stem cells destined to turn into glia or neurons. It can occur anywhere within the brain and can have multiple sites of origins, with a high probability of metastasis through cerebrospinal fluid (CSF). Metastases outside the central nervous system have been reported, but remain rare.