A brain tumor occurs when abnormal cells form within the brain. There are two main types of tumors: malignant (cancerous) tumors and benign (non-cancerous) tumors. These can be further classified as primary tumors, which start within the brain, and secondary tumors, which most commonly have spread from tumors located outside the brain, known as brain metastasis tumors. All types of brain tumors may produce symptoms that vary depending on the size of the tumor and the part of the brain that is involved. Where symptoms exist, they may include headaches, seizures, problems with vision, vomiting and mental changes. Other symptoms may include difficulty walking, speaking, with sensations, or unconsciousness.

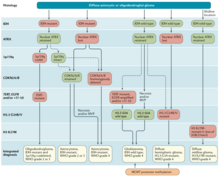
A glioma is a type of tumor that starts in the glial cells of the brain or the spine. Gliomas comprise about 30 percent of all brain tumors and central nervous system tumours, and 80 percent of all malignant brain tumours.

Glioblastoma, previously known as glioblastoma multiforme (GBM), is the most aggressive and most common type of cancer that originates in the brain, and has a very poor prognosis for survival. Initial signs and symptoms of glioblastoma are nonspecific. They may include headaches, personality changes, nausea, and symptoms similar to those of a stroke. Symptoms often worsen rapidly and may progress to unconsciousness.

Astrocytoma is a type of brain tumor. Astrocytomas originate from a specific kind of star-shaped glial cell in the cerebrum called an astrocyte. This type of tumor does not usually spread outside the brain and spinal cord and it does not usually affect other organs. After glioblastomas, astrocytomas are the second most common glioma and can occur in most parts of the brain and occasionally in the spinal cord.

Oligoastrocytomas are a subset of brain tumors that present with an appearance of mixed glial cell origin, astrocytoma and oligodendroglioma. However, the term "Oligoastrocytoma" is now considered obsolete by the National Comprehensive Cancer Network stating "the term should no longer be used as such morphologically ambiguous tumors can be reliably resolved into astrocytomas and oligodendrogliomas with molecular testing."

A blastoma is a type of cancer, more common in children, that is caused by malignancies in precursor cells, often called blasts. Examples are nephroblastoma, medulloblastoma, and retinoblastoma. The suffix -blastoma is used to imply a tumor of primitive, incompletely differentiated cells, e.g., chondroblastoma is composed of cells resembling the precursor of chondrocytes.

A ganglioglioma is a rare, slow-growing primary central nervous system (CNS) tumor which most frequently occurs in the temporal lobes of children and young adults. They are mixed cell tumors containing both neural ganglionic cells and neural glial cell components.

A gemistocyte is a swollen, reactive astrocyte.
Glial tumor is a general term for numerous tumors of the central nervous system, including astrocytomas, ependymal tumors, Oligodendroglioma, and primitive neuroectodermal tumors. The World Health Organization (WHO) classifies tumors into different categories according to severity and recurrence.The first tumor classified as grade I is called pilocytic astrocytoma and it is most commonly observed in children rather than adults. The next tumor is never common in the Dns called diffuse astrocytoma and it is considered a grade II, they are benign, or noncancerous, but can become malignant, meaning cancerous, as the tumor progresses. Grades III and grade IV are considered malignant astrocytomas. Anaplastic astrocytomas are considered by the WHO to be a grade III astrocytoma and Glioblastoma is a grade IV both are referred to high-grade glial tumors.

Gliosarcoma is a rare type of glioma, a cancer of the brain that comes from glial, or supportive, brain cells, as opposed to the neural brain cells. Gliosarcoma is a malignant cancer, and is defined as a glioblastoma consisting of gliomatous and sarcomatous components. Primary gliosarcoma (PGS) is classified as a grade IV tumor and a subtype of glioblastoma multiforme in the 2007 World Health Organization classification system (GBM). Because of a lack of specific and clear diagnostic criteria, the word "gliosarcoma" was frequently used to refer to glial tumours with mesenchymal properties, such as the ability to make collagen and reticulin.
Virtual karyotype is the digital information reflecting a karyotype, resulting from the analysis of short sequences of DNA from specific loci all over the genome, which are isolated and enumerated. It detects genomic copy number variations at a higher resolution for level than conventional karyotyping or chromosome-based comparative genomic hybridization (CGH). The main methods used for creating virtual karyotypes are array-comparative genomic hybridization and SNP arrays.

The giant-cell glioblastoma is a histological variant of glioblastoma, presenting a prevalence of bizarre, multinucleated giant cells.
Neuro-oncology is the study of brain and spinal cord neoplasms, many of which are very dangerous and life-threatening. Among the malignant brain cancers, gliomas of the brainstem and pons, glioblastoma multiforme, and high-grade astrocytoma/oligodendroglioma are among the worst. In these cases, untreated survival usually amounts to only a few months, and survival with current radiation and chemotherapy treatments may extend that time from around a year to a year and a half, possibly two or more, depending on the patient's condition, immune function, treatments used, and the specific type of malignant brain neoplasm. Surgery may in some cases be curative, but, as a general rule, malignant brain cancers tend to regenerate and emerge from remission easily, especially highly malignant cases. In such cases, the goal is to excise as much of the mass and as much of the tumor margin as possible without endangering vital functions or other important cognitive abilities. The Journal of Neuro-Oncology is the longest continuously published journal in the field and serves as a leading reference to those practicing in the area of neuro-oncology.

Anaplastic astrocytoma is a rare WHO grade III type of astrocytoma, which is a type of cancer of the brain. In the United States, the annual incidence rate for anaplastic astrocytoma is 0.44 per 100,000 people.

Temozolomide, sold under the brand name Temodar among others, is an anticancer medication used to treat brain tumors such as glioblastoma and anaplastic astrocytoma. It is taken by mouth or via intravenous infusion.

Isocitrate dehydrogenase 1 (NADP+), soluble is an enzyme that in humans is encoded by the IDH1 gene on chromosome 2. Isocitrate dehydrogenases catalyze the oxidative decarboxylation of isocitrate to 2-oxoglutarate. These enzymes belong to two distinct subclasses, one of which uses NAD+ as the electron acceptor and the other NADP+. Five isocitrate dehydrogenases have been reported: three NAD+-dependent isocitrate dehydrogenases, which localize to the mitochondrial matrix, and two NADP+-dependent isocitrate dehydrogenases, one of which is mitochondrial and the other predominantly cytosolic. Each NADP+-dependent isozyme is a homodimer. The protein encoded by this gene is the NADP+-dependent isocitrate dehydrogenase found in the cytoplasm and peroxisomes. It contains the PTS-1 peroxisomal targeting signal sequence. The presence of this enzyme in peroxisomes suggests roles in the regeneration of NADPH for intraperoxisomal reductions, such as the conversion of 2,4-dienoyl-CoAs to 3-enoyl-CoAs, as well as in peroxisomal reactions that consume 2-oxoglutarate, namely the alpha-hydroxylation of phytanic acid. The cytoplasmic enzyme serves a significant role in cytoplasmic NADPH production. Alternatively spliced transcript variants encoding the same protein have been found for this gene. [provided by RefSeq, Sep 2013]
Alternating electric field therapy, sometimes called tumor treating fields (TTFields), is a type of electromagnetic field therapy using low-intensity, intermediate frequency electrical fields to treat cancer. TTFields disrupt cell division by disrupting dipole alignment and inducing dielectrophoresis of critical molecules and organelles during mitosis. These anti-mitotic effects lead to cell death, slowing cancer growth. A TTField-treatment device manufactured by the Israeli company Novocure is approved in the United States and Europe for the treatment of newly diagnosed and recurrent glioblastoma, malignant pleural mesothelioma (MPM), and is undergoing clinical trials for several other tumor types. Despite earning regulatory approval, the efficacy of this technology remains controversial among medical experts.

Anaplastic oligodendroglioma is a neuroepithelial tumor which is believed to originate from oligodendrocytes, a cell type of the glia. In the World Health Organization (WHO) classification of brain tumors, anaplastic oligodendrogliomas are classified as grade III. In the course of the disease, it can degenerate into highly malignant oligodendroglioma, grade IV. The vast majority of oligodendrogliomas occur sporadically, without a confirmed cause and without inheritance within a family.
Diffuse leptomeningeal glioneuronal tumor (DLGNT) is a rare, primary CNS tumor, classified as distinct entity in 2016 and described as diffuse oligodendroglial-like leptomeningeal tumor of children in the 2016 classification of CNS neoplasms by the WHO., Typically, it's considered juvenile tumors but can occur in adults, the average age of diagnosis is five years. It's characterised by wide leptomeningeal spread with male predominance, like histopathology of neurocytoma, oligodendrocyte-like cytopathology, bland appearance, and severe clinical behaviour. Children's basal cisterns and inter-hemispheric fissures are typically involved in plaque like subarachnoid tumors. A common related intraparenchymal lesion is a spinal lesion. However, in certain situations, superficial parenchyma or Virchow-Robin gaps were affected.
Molecular and genetic investigations frequently show a combination of KIAA1549 and the serine/threonine protein kinase BRAF gene, and also deletions of the short arm of chromosome number 1 and/or the long arm of chromosome number 19.
Thalamic gliomas are very rare, deep-seated, generally high-grade glial neoplasms that form in the thalamus, representing 1–5% of all pediatric brain tumors. Because of their difficult to reach position, they are a unique and difficult challenge for neuro-oncologists and neurosurgeons.