
Electrochemistry is the branch of physical chemistry concerned with the relationship between electrical potential difference, as a measurable and quantitative phenomenon, and identifiable chemical change, with the potential difference as an outcome of a particular chemical change, or vice versa. These reactions involve electrons moving via an electronically-conducting phase between electrodes separated by an ionically conducting and electronically insulating electrolyte.

A scanning tunneling microscope (STM) is a type of microscope used for imaging surfaces at the atomic level. Its development in 1981 earned its inventors, Gerd Binnig and Heinrich Rohrer, then at IBM Zürich, the Nobel Prize in Physics in 1986. STM senses the surface by using an extremely sharp conducting tip that can distinguish features smaller than 0.1 nm with a 0.01 nm depth resolution. This means that individual atoms can routinely be imaged and manipulated. Most microscopes are built for use in ultra-high vacuum at temperatures approaching zero kelvin, but variants exist for studies in air, water and other environments, and for temperatures over 1000 °C.
In electrochemistry, the Nernst equation is a chemical thermodynamical relationship that permits the calculation of the reduction potential of a reaction from the standard electrode potential, absolute temperature, the number of electrons involved in the oxydo-reduction reaction, and activities of the chemical species undergoing reduction and oxidation respectively. It was named after Walther Nernst, a German physical chemist who formulated the equation.
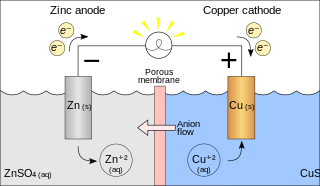
A galvanic cell or voltaic cell, named after the scientists Luigi Galvani and Alessandro Volta, respectively, is an electrochemical cell in which an electric current is generated from spontaneous Oxidation-Reduction reactions. A common apparatus generally consists of two different metals, each immersed in separate beakers containing their respective metal ions in solution that are connected by a salt bridge or separated by a porous membrane.

Cyclic voltammetry (CV) is a type of potentiodynamic electrochemical measurement. In a cyclic voltammetry experiment, the working electrode potential is ramped linearly versus time. Unlike in linear sweep voltammetry, after the set potential is reached in a CV experiment, the working electrode's potential is ramped in the opposite direction to return to the initial potential. These cycles of ramps in potential may be repeated as many times as needed. The current at the working electrode is plotted versus the applied voltage to give the cyclic voltammogram trace. Cyclic voltammetry is generally used to study the electrochemical properties of an analyte in solution or of a molecule that is adsorbed onto the electrode.

Chronoamperometry is an electrochemical technique in which the potential of the working electrode is stepped and the resulting current from faradaic processes occurring at the electrode is monitored as a function of time. The functional relationship between current response and time is measured after applying single or double potential step to the working electrode of the electrochemical system. Limited information about the identity of the electrolyzed species can be obtained from the ratio of the peak oxidation current versus the peak reduction current. However, as with all pulsed techniques, chronoamperometry generates high charging currents, which decay exponentially with time as any RC circuit. The Faradaic current - which is due to electron transfer events and is most often the current component of interest - decays as described in the Cottrell equation. In most electrochemical cells this decay is much slower than the charging decay-cells with no supporting electrolyte are notable exceptions. Most commonly a three electrode system is used. Since the current is integrated over relatively longer time intervals, chronoamperometry gives a better signal to noise ratio in comparison to other amperometric techniques.
In electrochemistry, overpotential is the potential difference (voltage) between a half-reaction's thermodynamically determined reduction potential and the potential at which the redox event is experimentally observed. The term is directly related to a cell's voltage efficiency. In an electrolytic cell the existence of overpotential implies that the cell requires more energy than thermodynamically expected to drive a reaction. In a galvanic cell the existence of overpotential means less energy is recovered than thermodynamics predicts. In each case the extra/missing energy is lost as heat. The quantity of overpotential is specific to each cell design and varies across cells and operational conditions, even for the same reaction. Overpotential is experimentally determined by measuring the potential at which a given current density is achieved.
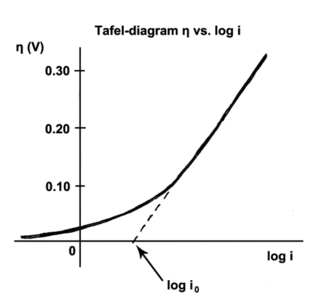
The Tafel equation is an equation in electrochemical kinetics relating the rate of an electrochemical reaction to the overpotential. The Tafel equation was first deduced experimentally and was later shown to have a theoretical justification. The equation is named after Swiss chemist Julius Tafel.
" It describes how the electrical current through an electrode depends on the voltage difference between the electrode and the bulk electrolyte for a simple, unimolecular redox reaction ".
Electroanalytical methods are a class of techniques in analytical chemistry which study an analyte by measuring the potential (volts) and/or current (amperes) in an electrochemical cell containing the analyte. These methods can be broken down into several categories depending on which aspects of the cell are controlled and which are measured. The three main categories are potentiometry, coulometry, and voltammetry.
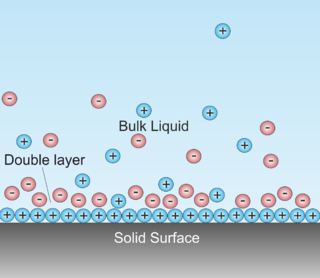
A double layer is a structure that appears on the surface of an object when it is exposed to a fluid. The object might be a solid particle, a gas bubble, a liquid droplet, or a porous body. The DL refers to two parallel layers of charge surrounding the object. The first layer, the surface charge, consists of ions adsorbed onto the object due to chemical interactions. The second layer is composed of ions attracted to the surface charge via the Coulomb force, electrically screening the first layer. This second layer is loosely associated with the object. It is made of free ions that move in the fluid under the influence of electric attraction and thermal motion rather than being firmly anchored. It is thus called the "diffuse layer".
In chemistry, an electrochemical reaction mechanism is the step by step sequence of elementary steps, involving at least one outer sphere electron transfer, by which an overall chemical change occurs.
A rotating disk electrode (RDE) is a working electrode used in three electrode systems for hydrodynamic voltammetry. The electrode rotates during experiments inducing a flux of analyte to the electrode. These working electrodes are used in electrochemical studies when investigating reaction mechanisms related to redox chemistry, among other chemical phenomena. The more complex rotating ring-disk electrode can be used as a rotating disk electrode if the ring is left inactive during the experiment.
An ultramicroelectrode (UME) is a working electrode used in a voltammetry. The small size of UME give them large diffusion layers and small overall currents. These features allow UME to achieve useful steady-state conditions and very high scan rates (V/s) with limited distortion. UME were developed independently by Wightman and Fleischmann around 1980. Small current at UME enables electrochemical measurements in low conductive media, where voltage drop associated with high solution resistance makes these experiments difficult for conventional electrodes. Furthermore, small voltage drop at UME leads to a very small voltage distortion at the electrode-solution interface which allows using two-electrode setup in voltammetric experiment instead of conventional three-electrode setup.
In electrochemistry, ITIES is an electrochemical interface that is either polarisable or polarised. An ITIES is polarisable if one can change the Galvani potential difference, or in other words the difference of inner potentials between the two adjacent phases, without noticeably changing the chemical composition of the respective phases. An ITIES system is polarised if the distribution of the different charges and redox species between the two phases determines the Galvani potential difference.
In electrochemistry, exchange current density is a parameter used in the Tafel equation, Butler–Volmer equation and other electrochemical kinetics expressions. The Tafel equation describes the dependence of current for an electrolytic process to overpotential.
Charge transfer coefficient, and symmetry factor are two related parameters used in description of the kinetics of electrochemical reactions. They appear in the Butler–Volmer equation and related expressions.
In cyclic voltammetry, the Randles–Ševčík equation describes the effect of scan rate on the peak current ip. For simple redox events such as the ferrocene/ferrocenium couple, ip depends not only on the concentration and diffusional properties of the electroactive species but also on scan rate.
Scanning electrochemical microscopy (SECM) is a technique within the broader class of scanning probe microscopy (SPM) that is used to measure the local electrochemical behavior of liquid/solid, liquid/gas and liquid/liquid interfaces. Initial characterization of the technique was credited to University of Texas electrochemist, Allen J. Bard, in 1989. Since then, the theoretical underpinnings have matured to allow widespread use of the technique in chemistry, biology and materials science. Spatially resolved electrochemical signals can be acquired by measuring the current at an ultramicroelectrode (UME) tip as a function of precise tip position over a substrate region of interest. Interpretation of the SECM signal is based on the concept of diffusion-limited current. Two-dimensional raster scan information can be compiled to generate images of surface reactivity and chemical kinetics.

Fast-scan cyclic voltammetry (FSCV) is cyclic voltammetry with a very high scan rate (up to 1×106 V·s−1). Application of high scan rate allows rapid acquisition of a voltammogram within several milliseconds and ensures high temporal resolution of this electroanalytical technique. An acquisition rate of 10 Hz is routinely employed.

Pseudocapacitance is the electrochemical storage of electricity in an electrochemical capacitor (Pseudocapacitor). This faradaic charge transfer originates by a very fast sequence of reversible faradaic redox, electrosorption or intercalation processes on the surface of suitable electrodes. Pseudocapacitance is accompanied by an electron charge-transfer between electrolyte and electrode coming from a de-solvated and adsorbed ion. One electron per charge unit is involved. The adsorbed ion has no chemical reaction with the atoms of the electrode since only a charge-transfer takes place.



























