
Ehlers–Danlos syndromes (EDS) are a group of 13 genetic connective-tissue disorders. Symptoms often include loose joints, joint pain, stretchy velvety skin, and abnormal scar formation. These may be noticed at birth or in early childhood. Complications may include aortic dissection, joint dislocations, scoliosis, chronic pain, or early osteoarthritis. The current classification was last updated in 2017, when a number of rarer forms of EDS were added.
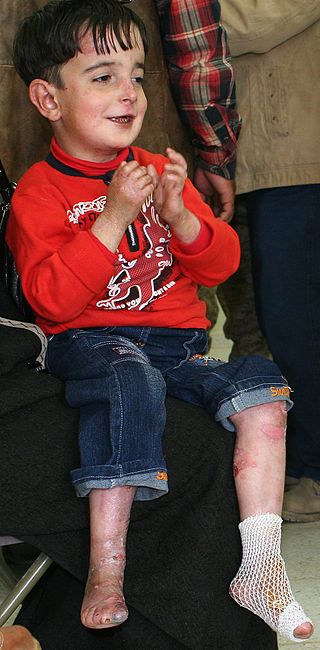
Epidermolysis bullosa (EB) is a group of rare medical conditions that result in easy blistering of the skin and mucous membranes. Blisters occur with minor trauma or friction and are painful. Its severity can range from mild to fatal. Inherited EB is a rare disease with a prevalence in the United States of 8.2 per million live births. Those with mild cases may not develop symptoms until they start to crawl or walk. Complications may include esophageal narrowing, squamous cell skin cancer, and the need for amputations.

Elastin is a protein that in humans is encoded by the ELN gene. Elastin is a key component of the extracellular matrix in gnathostomes. It is highly elastic and present in connective tissue allowing many tissues in the body to resume their shape after stretching or contracting. Elastin helps skin to return to its original position when it is poked or pinched. Elastin is also an important load-bearing tissue in the bodies of vertebrates and used in places where mechanical energy is required to be stored.
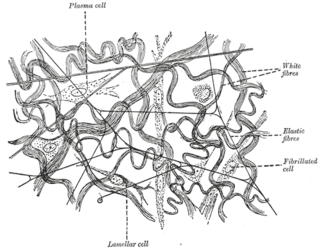
Elastic fibers are an essential component of the extracellular matrix composed of bundles of proteins (elastin) which are produced by a number of different cell types including fibroblasts, endothelial, smooth muscle, and airway epithelial cells. These fibers are able to stretch many times their length, and snap back to their original length when relaxed without loss of energy. Elastic fibers include elastin, elaunin and oxytalan.

Multidrug resistance-associated protein 6 (MRP6) also known as ATP-binding cassette sub-family C member 6 (ABCC6) and multi-specific organic anion transporter E (MOAT-E) is a protein that in humans is encoded by the ABCC6 gene. The protein encoded by the ABCC6 gene is a member of the superfamily of ATP-binding cassette (ABC) transporters.

Kindler syndrome is a rare congenital disease of the skin caused by a mutation in the KIND1 gene.

Genetic Alliance is a nonprofit organization, founded in 1986 by Joan O. Weiss, working with Victor A. McKusick, to advocate for health benefits in the accelerating field of genomic research. This organization is a network of over 1,000 disease advocacy organizations, universities, government organizations, private companies, and public policy organizations. They aim to advance genetic research agendas toward health benefit by engaging a broad range of stakeholders, including healthcare providers, researchers, industry professionals, public policy leaders, as well as individuals, families and communities. They create programs using a collaborative approach, and aim to increase efficiency and reduce obstacles in genetic research, while ensuring that voices from the involved disease communities are heard. They also promote public policies to advance healthcare. Genetic Alliance provides technical support and informational resources to guide disease-specific advocacy organizations in being their own research advocates. They also maintain a biobank as a central storage facility for several organizations who otherwise would not have the infrastructure to maintain their own repository.

Keratin 5, also known as KRT5, K5, or CK5, is a protein that is encoded in humans by the KRT5 gene. It dimerizes with keratin 14 and forms the intermediate filaments (IF) that make up the cytoskeleton of basal epithelial cells. This protein is involved in several diseases including epidermolysis bullosa simplex and breast and lung cancers.

Ablepharon macrostomia syndrome (AMS) is an extremely rare, autosomal dominant genetic disorder characterized by abnormal phenotypic appearances that primarily affect the head and face as well as the skull, skin, fingers and genitals. AMS generally results in abnormal ectoderm-derived structures. The most prominent abnormality is the underdevelopment (microblepharon) or absence of eyelids – signifying the ablepharon aspect of the disease – and a wide, fish-like mouth – macrostomia. Recent scholars and surgeons have called into question the naming of the condition as "Ablepharon" on account of recent investigation and histology showing consistent evidence of at least some eyelid tissue. Infants presenting with AMS may also have malformations of the abdominal wall and nipples. Children with AMS might also experience issues with learning development, language difficulties and intellectual disabilities.

Epidermolysis bullosa dystrophica or dystrophic EB (DEB) is an inherited disease affecting the skin and other organs.
Buschke–Ollendorff syndrome (BOS) is a rare genetic skin disorder associated with LEMD3 that typically presents with widespread painless papules.

Collagen alpha-1(VII) chain is a protein that in humans is encoded by the COL7A1 gene. It is composed of a triple helical, collagenous domain flanked by two non-collagenous domains, and functions as an anchoring fibril between the dermal-epidermal junction in the basement membrane. Mutations in COL7A1 cause all types of dystrophic epidermolysis bullosa, and the exact mutations vary based on the specific type or subtype. It has been shown that interactions between the NC-1 domain of collagen VII and several other proteins, including laminin-5 and collagen IV, contribute greatly to the overall stability of the basement membrane.

Dystonin(DST), also known as bullous pemphigoid antigen 1 (BPAG1), isoforms 1/2/3/4/5/8, is a protein that in humans is encoded by the DST gene.

Myosin-11 is a protein that in humans is encoded by the MYH11 gene.
Nodal modulator 1 is a protein that in humans is encoded by the NOMO1 gene.

Urbach–Wiethe disease is a very rare recessive genetic disorder, with approximately 400 reported cases since its discovery. It was first officially reported in 1929 by Erich Urbach and Camillo Wiethe, although cases may be recognized dating back as early as 1908.
Congenital generalized lipodystrophy is an extremely rare autosomal recessive condition, characterized by an extreme scarcity of fat in the subcutaneous tissues. It is a type of lipodystrophy disorder where the magnitude of fat loss determines the severity of metabolic complications. Only 250 cases of the condition have been reported, and it is estimated that it occurs in 1 in 10 million people worldwide.
Generalized arterial calcification of infancy (GACI) is an extremely rare genetic disorder. It is caused by mutations in the ENPP1 gene in 75% of the subjects or in mutations in the ABCC6 genes in 10% of patients. However, sometimes individuals affected with GACI do not have mutations in the ENPP1 or ABCC6 gene and in those cases the cause of the disorder is unknown.

Arterial calcification due to deficiency of CD73 (ACDC) is a rare genetic disorder that causes calcium buildup in the arteries and joints of the hands and feet, and other areas below the waist. Although patients exhibiting these symptoms have been identified as early as 1914, this disorder had not been studied extensively until recently. The identification of the specific ACDC gene and mutations occurred in 2011. ACDC is caused by a mutation in the NT5E gene, which prevents calcium-removing agents from functioning,. Patients with this mutation experience chronic pain, difficulty moving, and increased risk of cardiovascular problems. In experiments at the molecular level, treatment with adenosine or a phosphatase inhibitor reversed and prevented calcification, suggesting they could be used as possible treatment methods. There is currently no cure for ACDC, and patients have limited treatment options which focus primarily on removal of blood calcium and improving mobility.
INZ-701 is a recombinant ENPP1 enzyme developed to treat some genetic disorders that prevent normal production of ENPP1. It is developed by Inozyme Pharma.






