
A genetic disorder is a health problem caused by one or more abnormalities in the genome. It can be caused by a mutation in a single gene (monogenic) or multiple genes (polygenic) or by a chromosomal abnormality. Although polygenic disorders are the most common, the term is mostly used when discussing disorders with a single genetic cause, either in a gene or chromosome. The mutation responsible can occur spontaneously before embryonic development, or it can be inherited from two parents who are carriers of a faulty gene or from a parent with the disorder. When the genetic disorder is inherited from one or both parents, it is also classified as a hereditary disease. Some disorders are caused by a mutation on the X chromosome and have X-linked inheritance. Very few disorders are inherited on the Y chromosome or mitochondrial DNA.

Turner syndrome (TS), also known as 45,X, or 45,X0, is a genetic disorder in which a female is partially or completely missing an X chromosome. Signs and symptoms vary among those affected. Often, a short and webbed neck, low-set ears, low hairline at the back of the neck, short stature, and swollen hands and feet are seen at birth. Typically, those affected do not develop menstrual periods or breasts without hormone treatment and are unable to have children without reproductive technology. Heart defects, diabetes, and low thyroid hormone occur in the disorder more frequently than average. Most people with Turner syndrome have normal intelligence; however, many have problems with spatial visualization that may be needed in order to learn mathematics. Vision and hearing problems also occur more often than average.

Macrocephaly is a condition in which circumference of the human head is abnormally large. It may be pathological or harmless, and can be a familial genetic characteristic. People diagnosed with macrocephaly will receive further medical tests to determine whether the syndrome is accompanied by particular disorders. Those with benign or familial macrocephaly are considered to have megalencephaly.

Uniparental disomy (UPD) occurs when a person receives two copies of a chromosome, or of part of a chromosome, from one parent and no copy from the other. UPD can be the result of heterodisomy, in which a pair of non-identical chromosomes are inherited from one parent or isodisomy, in which a single chromosome from one parent is duplicated. Uniparental disomy may have clinical relevance for several reasons. For example, either isodisomy or heterodisomy can disrupt parent-specific genomic imprinting, resulting in imprinting disorders. Additionally, isodisomy leads to large blocks of homozygosity, which may lead to the uncovering of recessive genes, a similar phenomenon seen in inbred children of consanguineous partners.

Weissenbacher–Zweymuller syndrome (WZS), also called Pierre-Robin syndrome with fetal chondrodysplasia, is an autosomal recessive congenital disorder, linked to mutations in the COL11A2 gene, which codes for the α2 strand of collagen type XI. It is a collagenopathy, types II and XI disorder. The condition was first characterized in 1964 by G. Weissenbacher and Ernst Zweymüller.
Renal agenesis is a medical condition in which one (unilateral) or both (bilateral) fetal kidneys fail to develop.

Arthrogryposis (AMC) describes congenital joint contracture in two or more areas of the body. It derives its name from Greek, literally meaning 'curving of joints'.

Chédiak–Higashi syndrome (CHS) is a rare autosomal recessive disorder that arises from a mutation of a lysosomal trafficking regulator protein, which leads to a decrease in phagocytosis. The decrease in phagocytosis results in recurrent pyogenic infections, albinism, and peripheral neuropathy.
Hyperimmunoglobulinemia E syndrome (HIES), of which the autosomal dominant form is called Job's syndrome or Buckley syndrome, is a heterogeneous group of immune disorders. Job's is also very rare at about 300 cases currently in the literature.

Dermatoglyphics is the scientific study of fingerprints, lines, mounts and shapes of hands, as distinct from the superficially similar pseudoscience of palmistry.
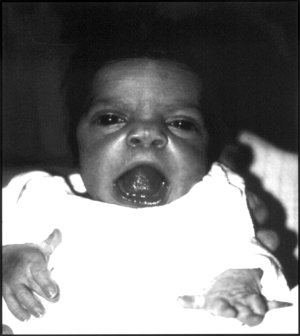
Robinow syndrome is an extremely rare genetic disorder characterized by short-limbed dwarfism, abnormalities in the head, face, and external genitalia, as well as vertebral segmentation. The disorder was first described in 1969 by human geneticist Meinhard Robinow, along with physicians Frederic N. Silverman and Hugo D. Smith, in the American Journal of Diseases of Children. By 2002, over 100 cases had been documented and introduced into medical literature.

Isodicentric 15, also called marker chromosome 15 syndrome, idic(15), partial tetrasomy 15q, or inverted duplication 15, is a chromosome abnormality in which a child is born with extra genetic material from chromosome 15. People with idic(15) are typically born with 47 chromosomes in their body cells, instead of the normal 46. The extra chromosome, which is classified as a small supernumerary marker chromosome, is made up of a piece of chromosome 15 that has been duplicated end-to-end like a mirror image. It is the presence of this extra genetic material that is thought to account for the symptoms seen in some people with idic(15). Individuals with idic(15) have a total of four copies of this chromosome 15 region instead of the usual two copies. The term isodicentric refers to a duplication and inversion of a centromere-containing chromosomal segment.
Lymphocyte function-associated antigen 1 (LFA-1) is an integrin found on lymphocytes and other leukocytes. LFA-1 plays a key role in emigration, which is the process by which leukocytes leave the bloodstream to enter the tissues. LFA-1 also mediates firm arrest of leukocytes. Additionally, LFA-1 is involved in the process of cytotoxic T cell mediated killing as well as antibody mediated killing by granulocytes and monocytes. As of 2007, LFA-1 has 6 known ligands: ICAM-1, ICAM-2, ICAM-3, ICAM-4, ICAM-5, and JAM-A. LFA-1/ICAM-1 interactions have recently been shown to stimulate signaling pathways that influence T cell differentiation. LFA-1 belongs to the integrin superfamily of adhesion molecules.
Congenital limb deformities are congenital musculoskeletal disorders which primarily affect the upper and lower limbs.

Persistent Fetal Vasculature(PFV), known also as Persistent Fetal Vasculature Syndrome (PFSV), and until 1997 known primarily as Persistent Hyperplastic Primary Vitreous (PHPV), is a rare congenital anomaly which occurs when blood vessels within the developing eye, known as the embryonic hyaloid vasculature network, fail to regress as they normally would in-utero after the eye is fully developed. Defects which arise from this lack of vascular regression are diverse; as a result, the presentation, symptoms, and prognosis of affected patients vary widely, ranging from clinical insignificance to irreversible blindness. The underlying structural causes of PFV are considered to be relatively common, and the vast majority of cases do not warrant additional intervention. When symptoms do manifest, however, they are often significant, causing detrimental and irreversible visual impairment. Persistent Fetal Vasculature heightens the lifelong risk of glaucoma, cataracts, intraocular hemorrhages, and retinal detachments, accounting for the visual loss of nearly 5% of the blind community in the developed world. In diagnosed cases of PFV, approximately 90% of patients with a unilateral disease have associated poor vision in the affected eye.

Pitt–Hopkins syndrome (PTHS) is a rare genetic disorder characterized by developmental delay, epilepsy, distinctive facial features, and possible intermittent hyperventilation followed by apnea. Pitt-Hopkins syndrome can be marked by intellectual disabilities as well as problems with socializing. It is part of the clinical spectrum of Rett-like syndromes.

Ectrodactyly, split hand, or cleft hand involves the deficiency or absence of one or more central digits of the hand or foot and is also known as split hand/split foot malformation (SHFM). The hands and feet of people with ectrodactyly (ectrodactyls) are often described as "claw-like" and may include only the thumb and one finger with similar abnormalities of the feet.
Wiedemann–Rautenstrauch (WR) syndrome, also known as neonatal progeroid syndrome, is a rare autosomal recessive progeroid syndrome. There have been over 30 cases of WR. WR is associated with abnormalities in bone maturation, and lipids and hormone metabolism.

Ring chromosome 15 is a condition that arises when chromosome 15 fuses to form a ring chromosome. Usually, ring chromosome 15 forms due to the modification or deletion of genetic information on chromosome 15 in the preliminary stages of embryonic development, but it can rarely also be inherited.
Compton-North congenital myopathy, also known as congenital lethal myopathy, Compton-North type, is a rare, fatal genetic disorder of pre-natal onset that results in death shortly after birth and is characterized by fetal akinesia and movement restriction, polyhydramnios, severe hypotonia of neonatal-onset, generalized weakness of the respiratory, bulbar, and skeletal muscles, presence of multiple congenital muscular contractures.



