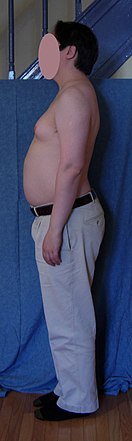
XYY syndrome, also known as Jacobs syndrome, is an aneuploid genetic condition in which a male has an extra Y chromosome. There are usually few symptoms. These may include being taller than average and an increased risk of learning disabilities. The person is generally otherwise normal, including typical rates of fertility.

Turner syndrome (TS), also known as 45,X, or 45,X0, is a genetic disorder in which a female is partially or completely missing an X chromosome. Most people have two sex chromosomes. It only affects females. Signs and symptoms vary among those affected. Often, a short and webbed neck, low-set ears, low hairline at the back of the neck, short stature, and swollen hands and feet are seen at birth. Typically, those affected do not develop menstrual periods or breasts without hormone treatment and are unable to have children without reproductive technology. Heart defects, diabetes, and hypothyroidism occur in the disorder more frequently than average. Most people with Turner syndrome have normal intelligence; however, many have problems with spatial visualization that may be needed in order to learn mathematics. Vision and hearing problems also occur more often than average.
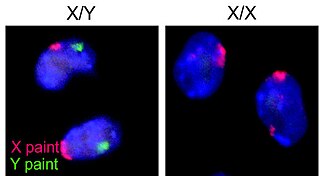
The XY sex-determination system is a sex-determination system used to classify many mammals, including humans, some insects (Drosophila), some snakes, some fish (guppies), and some plants. In this system, the sex of an individual is determined by a pair of sex chromosomes. Females have two of the same kind of sex chromosome (XX), and are called the homogametic sex. Males have two different kinds of sex chromosomes (XY), and are called the heterogametic sex.
Delayed puberty is when a person lacks or has incomplete development of specific sexual characteristics past the usual age of onset of puberty. The person may have no physical or hormonal signs that puberty has begun. In the United States, girls are considered to have delayed puberty if they lack breast development by age 13 or have not started menstruating by age 15. Boys are considered to have delayed puberty if they lack enlargement of the testicles by age 14. Delayed puberty affects about 2% of adolescents.
Hypogonadism means diminished functional activity of the gonads—the testicles or the ovaries—that may result in diminished production of sex hormones. Low androgen levels are referred to as hypoandrogenism and low estrogen as hypoestrogenism. These are responsible for the observed signs and symptoms in both males and females.
Kallmann syndrome (KS) is a genetic disorder that prevents a person from starting or fully completing puberty. Kallmann syndrome is a form of a group of conditions termed hypogonadotropic hypogonadism. To distinguish it from other forms of hypogonadotropic hypogonadism, Kallmann syndrome has the additional symptom of a total lack of sense of smell (anosmia) or a reduced sense of smell. If left untreated, people will have poorly defined secondary sexual characteristics, show signs of hypogonadism, almost invariably are infertile and are at increased risk of developing osteoporosis. A range of other physical symptoms affecting the face, hands and skeletal system can also occur.

XX male syndrome, also known as de la Chapelle syndrome, is a rare congenital intersex condition in which an individual with a 46,XX karyotype develops a male phenotype. Synonyms include 46,XX testicular difference of sex development, 46,XX sex reversal, nonsyndromic 46,XX testicular DSD, and XX sex reversal.

XXYY syndrome is a sex chromosome anomaly in which males have 2 extra chromosomes, one X and one Y chromosome. Human cells usually contain two sex chromosomes, one from the mother and one from the father. Usually, females have two X chromosomes (XX) and males have one X and one Y chromosome (XY). The appearance of at least one Y chromosome with a properly functioning SRY gene makes a male. Therefore, humans with XXYY are genotypically male. Males with XXYY syndrome have 48 chromosomes instead of the typical 46. This is why XXYY syndrome is sometimes written as 48, XXYY syndrome or 48, XXYY. It affects an estimated one in every 18,000–40,000 male births.

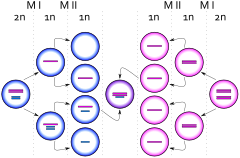
49,XXXXY syndrome is an extremely rare aneuploidic sex chromosomal abnormality. It occurs in approximately 1 out of 85,000 to 100,000 males. This syndrome is the result of maternal non-disjunction during both meiosis I and II. It was first diagnosed in 1960 and was coined Fraccaro syndrome after the researcher.

Gonadal dysgenesis is classified as any congenital developmental disorder of the reproductive system in humans. It is atypical development of gonads in an embryo. One type of gonadal dysgenesis is the development of functionless, fibrous tissue, termed streak gonads, instead of reproductive tissue. Streak gonads are a form of aplasia, resulting in hormonal failure that manifests as sexual infantism and infertility, with no initiation of puberty and secondary sex characteristics.
Disorders of sex development (DSDs), also known as differences in sex development or variations in sex characteristics (VSC), are congenital conditions affecting the reproductive system, in which development of chromosomal, gonadal, or anatomical sex is atypical.
Wilson-Turner syndrome (WTS), also known as mental retardation X linked syndromic 6 (MRXS6), and mental retardation X linked with gynecomastia and obesity is a congenital condition characterized by intellectual disability and associated with childhood-onset obesity. It is found to be linked to the X chromosome and caused by a mutation in the HDAC8 gene, which is located on the q arm at locus 13.1. Individuals with Wilson–Turner syndrome have a spectrum of physical characteristics including dysmorphic facial features, hypogonadism, and short stature. Females generally have milder phenotypes than males. This disorder affects all demographics equally and is seen in less than one in one million people.
Harry Fitch Klinefelter Jr. was an American rheumatologist and endocrinologist. Klinefelter syndrome is named after him.
Hypergonadotropic hypogonadism (HH), also known as primary or peripheral/gonadal hypogonadism or primary gonadal failure, is a condition which is characterized by hypogonadism which is due to an impaired response of the gonads to the gonadotropins, follicle-stimulating hormone (FSH) and luteinizing hormone (LH), and in turn a lack of sex steroid production. As compensation and the lack of negative feedback, gonadotropin levels are elevated. Individuals with HH have an intact and functioning hypothalamus and pituitary glands so they are still able to produce FSH and LH. HH may present as either congenital or acquired, but the majority of cases are of the former nature. HH can be treated with hormone replacement therapy.
XXXYsyndrome is a genetic condition characterized by a sex chromosome aneuploidy, where individuals have two extra X chromosomes. People in most cases have two sex chromosomes: an X and a Y or two X chromosomes. The presence of one Y chromosome with a functioning SRY gene causes the expression of genes that determine maleness. Because of this, XXXY syndrome only affects males. The additional two X chromosomes in males with XXXY syndrome causes them to have 48 chromosomes, instead of the typical 46. XXXY syndrome is therefore often referred to as 48,XXXY. There is a wide variety of symptoms associated with this syndrome, including cognitive and behavioral problems, taurodontism, and infertility. This syndrome is usually inherited via a new mutation in one of the parents' gametes, as those affected by it are usually infertile. It is estimated that XXXY affects one in every 50,000 male births.

Tetrasomy X, also known as 48,XXXX, is a chromosomal disorder in which a female has four, rather than two, copies of the X chromosome. It is associated with intellectual disability of varying severity, characteristic "coarse" facial features, heart defects, and skeletal anomalies such as increased height, clinodactyly, and radioulnar synostosis. Tetrasomy X is a rare condition, with few medically recognized cases; it is estimated to occur in approximately 1 in 50,000 females.
Sexual anomalies, also known as sexual abnormalities, are a set of clinical conditions due to chromosomal, gonadal and/or genitalia variation. Individuals with congenital (inborn) discrepancy between sex chromosome, gonadal, and their internal and external genitalia are categorised as individuals with a disorder of sex development (DSD). Afterwards, if the family or individual wishes, they can partake in different management and treatment options for their conditions.

Pentasomy X, also known as 49,XXXXX, is a chromosomal disorder in which a female has five, rather than two, copies of the X chromosome. Pentasomy X is associated with short stature, intellectual disability, characteristic facial features, heart defects, skeletal anomalies, and pubertal and reproductive abnormalities. The condition is exceptionally rare, with an estimated prevalence between 1 in 85,000 and 1 in 250,000.

Trisomy X, also known as triple X syndrome and characterized by the karyotype 47,XXX, is a chromosome disorder in which a female has an extra copy of the X chromosome. It is relatively common and occurs in 1 in 1,000 females, but is rarely diagnosed; fewer than 10% of those with the condition know they have it.

XXXYY syndrome, also known as 49,XXXYY, is a chromosomal disorder in which a male has three copies of the X chromosome and two copies of the Y chromosome. XXXYY syndrome is exceptionally rare, with only eight recorded cases. Little is known about its presentation, but associated characteristics include intellectual disability, anomalies of the external genitalia, and characteristic physical and facial features. It is not caused by characteristics of the parents, but rather occurs via nondisjunction, a random event in gamete development. The karyotype observed in the syndrome is formally known as 49,XXXYY, which represents the 49 chromosomes observed in the disorder as compared to the 46 in normal human development.