Hydrogenation is a chemical reaction between molecular hydrogen (H2) and another compound or element, usually in the presence of a catalyst such as nickel, palladium or platinum. The process is commonly employed to reduce or saturate organic compounds. Hydrogenation typically constitutes the addition of pairs of hydrogen atoms to a molecule, often an alkene. Catalysts are required for the reaction to be usable; non-catalytic hydrogenation takes place only at very high temperatures. Hydrogenation reduces double and triple bonds in hydrocarbons.

BINAP (2,2′-bis(diphenylphosphino)-1,1′-binaphthyl) is an organophosphorus compound. This chiral diphosphine ligand is widely used in asymmetric synthesis. It consists of a pair of 2-diphenylphosphinonaphthyl groups linked at the 1 and 1′ positions. This C2-symmetric framework lacks a stereogenic atom, but has axial chirality due to restricted rotation (atropisomerism). The barrier to racemization is high due to steric hindrance, which limits rotation about the bond linking the naphthyl rings. The dihedral angle between the naphthyl groups is approximately 90°. The natural bite angle is 93°.
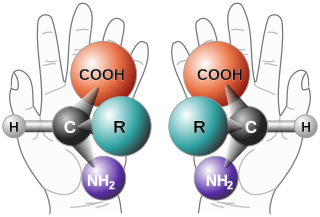
Enantioselective synthesis, also called asymmetric synthesis, is a form of chemical synthesis. It is defined by IUPAC as "a chemical reaction in which one or more new elements of chirality are formed in a substrate molecule and which produces the stereoisomeric products in unequal amounts."

In organic chemistry, the Michael reaction or Michael 1,4 addition is a reaction between a Michael donor and a Michael acceptor to produce a Michael adduct by creating a carbon-carbon bond at the acceptor's β-carbon. It belongs to the larger class of conjugate additions and is widely used for the mild formation of carbon-carbon bonds.
Reductive amination is a form of amination that involves the conversion of a carbonyl group to an amine via an intermediate imine. The carbonyl group is most commonly a ketone or an aldehyde. It is a common method to make amines and is widely used in green chemistry since it can be done catalytically in one-pot under mild conditions. In biochemistry, dehydrogenase enzymes use reductive amination to produce the amino acid, glutamate. Additionally, there is ongoing research on alternative synthesis mechanisms with various metal catalysts which allow the reaction to be less energy taxing, and require milder reaction conditions. Investigation into biocatalysts, such as imine reductases, have allowed for higher selectivity in the reduction of chiral amines which is an important factor in pharmaceutical synthesis.

The Corey–Itsuno reduction, also known as the Corey–Bakshi–Shibata (CBS) reduction, is a chemical reaction in which a prochiral ketone is enantioselectively reduced to produce the corresponding chiral, non-racemic alcohol. The oxazaborolidine reagent which mediates the enantioselective reduction of ketones was previously developed by the laboratory of Itsuno and thus this transformation may more properly be called the Itsuno-Corey oxazaborolidine reduction.
The Strecker amino acid synthesis, also known simply as the Strecker synthesis, is a method for the synthesis of amino acids by the reaction of an aldehyde with cyanide in the presence of ammonia. The condensation reaction yields an α-aminonitrile, which is subsequently hydrolyzed to give the desired amino acid. The method is used for the commercial production of racemic methionine from methional.
The Meerwein–Ponndorf–Verley (MPV) reduction in organic chemistry is the reduction of ketones and aldehydes to their corresponding alcohols utilizing aluminium alkoxide catalysis in the presence of a sacrificial alcohol. The advantages of the MPV reduction lie in its high chemoselectivity, and its use of a cheap environmentally friendly metal catalyst. MPV reductions have been described as "obsolete" owing to the development of sodium borohydride and related reagents.
In organic chemistry, kinetic resolution is a means of differentiating two enantiomers in a racemic mixture. In kinetic resolution, two enantiomers react with different reaction rates in a chemical reaction with a chiral catalyst or reagent, resulting in an enantioenriched sample of the less reactive enantiomer. As opposed to chiral resolution, kinetic resolution does not rely on different physical properties of diastereomeric products, but rather on the different chemical properties of the racemic starting materials. The enantiomeric excess (ee) of the unreacted starting material continually rises as more product is formed, reaching 100% just before full completion of the reaction. Kinetic resolution relies upon differences in reactivity between enantiomers or enantiomeric complexes.

In organic chemistry, organocatalysis is a form of catalysis in which the rate of a chemical reaction is increased by an organic catalyst. This "organocatalyst" consists of carbon, hydrogen, sulfur and other nonmetal elements found in organic compounds. Because of their similarity in composition and description, they are often mistaken as a misnomer for enzymes due to their comparable effects on reaction rates and forms of catalysis involved.
The Hajos–Parrish–Eder–Sauer–Wiechert reaction in organic chemistry is a proline catalysed asymmetric aldol reaction. The reaction is named after the principal investigators of the two groups who reported it simultaneously: Zoltan Hajos and David Parrish from Hoffmann-La Roche and Rudolf Wiechert and co-workers from Schering AG. Discovered in the 1970s the original Hajos-Parrish catalytic procedure – shown in the reaction equation, leading to the optically active bicyclic ketol – paved the way of asymmetric organocatalysis. The Eder-Sauer-Wiechert modification lead directly to the optically active enedione, through the loss of water from the bicyclic ketol shown in figure.
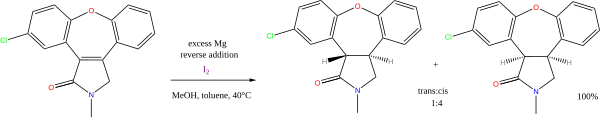
Asymmetric hydrogenation is a chemical reaction that adds two atoms of hydrogen to a target (substrate) molecule with three-dimensional spatial selectivity. Critically, this selectivity does not come from the target molecule itself, but from other reagents or catalysts present in the reaction. This allows spatial information to transfer from one molecule to the target, forming the product as a single enantiomer. The chiral information is most commonly contained in a catalyst and, in this case, the information in a single molecule of catalyst may be transferred to many substrate molecules, amplifying the amount of chiral information present. Similar processes occur in nature, where a chiral molecule like an enzyme can catalyse the introduction of a chiral centre to give a product as a single enantiomer, such as amino acids, that a cell needs to function. By imitating this process, chemists can generate many novel synthetic molecules that interact with biological systems in specific ways, leading to new pharmaceutical agents and agrochemicals. The importance of asymmetric hydrogenation in both academia and industry contributed to two of its pioneers — William Standish Knowles and Ryōji Noyori — being collectively awarded one half of the 2001 Nobel Prize in Chemistry.
Within the area of organocatalysis, (thio)urea organocatalysis describes the use of ureas and thioureas to accelerate and stereochemically alter organic transformations. The effects arise through hydrogen-bonding interactions between the substrate and the (thio)urea. Unlike classical catalysts, these organocatalysts interact by non-covalent interactions, especially hydrogen bonding. The scope of these small-molecule H-bond donors termed (thio)urea organocatalysis covers both non-stereoselective and stereoselective applications.
Enantioselective ketone reductions convert prochiral ketones into chiral, non-racemic alcohols and are used heavily for the synthesis of stereodefined alcohols.

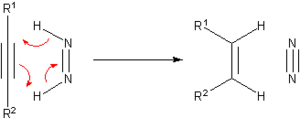
In chemistry, the hydrogenation of carbon–nitrogen double bonds is the addition of the elements of dihydrogen (H2) across a carbon–nitrogen double bond, forming amines or amine derivatives. Although a variety of general methods have been developed for the enantioselective hydrogenation of ketones, methods for the hydrogenation of carbon–nitrogen double bonds are less general. Hydrogenation of imines is complicated by both syn/anti isomerization and tautomerization to enamines, which may be hydrogenated with low enantioselectivity in the presence of a chiral catalyst. Additionally, the substituent attached to nitrogen affects both the reactivity and spatial properties of the imine, complicating the development of a general catalyst system for imine hydrogenation. Despite these challenges, methods have been developed that address particular substrate classes, such as N-aryl, N-alkyl, and endocyclic imines.

Hydrogen-bond catalysis is a type of organocatalysis that relies on use of hydrogen bonding interactions to accelerate and control organic reactions. In biological systems, hydrogen bonding plays a key role in many enzymatic reactions, both in orienting the substrate molecules and lowering barriers to reaction. However, chemists have only recently attempted to harness the power of using hydrogen bonds to perform catalysis, and the field is relatively undeveloped compared to research in Lewis acid catalysis.
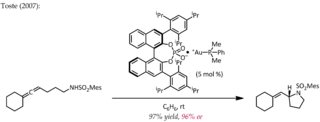
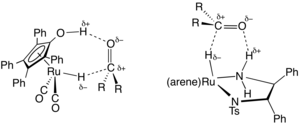
Asymmetric counteranion directed catalysis (ACDC) or chiral anion catalysis in enantioselective synthesis is the "induction of enantioselectivity in a reaction proceeding through a cationic intermediate by means of ion pairing with a chiral, enantiomerically pure anion provided by the catalyst". Although chiral Brønsted acid catalyzed reactions may well fall into this category of catalysis under the definition given here, the extent of proton transfer and the demarcation between hydrogen bonding and full proton transfer is often ambiguous. Hence, some authors may exclude ion pair formation by proton transfer as a type of chiral counteranion catalysis. The discussion below will focus on chiral ion pairs generated through means other than proton transfer.
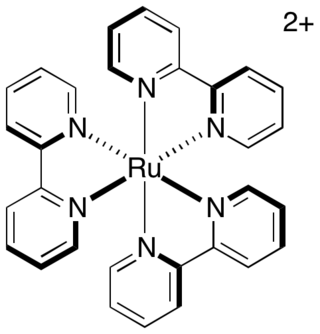
Photoredox catalysis is a branch of photochemistry that uses single-electron transfer. Photoredox catalysts are generally drawn from three classes of materials: transition-metal complexes, organic dyes, and semiconductors. While organic photoredox catalysts were dominant throughout the 1990s and early 2000s, soluble transition-metal complexes are more commonly used today.

The A3 coupling (also known as A3 coupling reaction or the aldehyde-alkyne-amine reaction), coined by Prof. Chao-Jun Li of McGill University, is a type of multicomponent reaction involving an aldehyde, an alkyne and an amine which react to give a propargylamine.
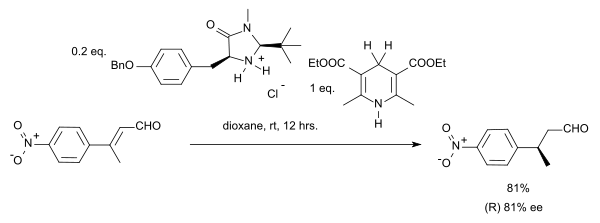
The ketimine Mannich reaction is an asymmetric synthetic technique using differences in starting material to push a Mannich reaction to create an enantiomeric product with steric and electronic effects, through the creation of a ketimine group. Typically, this is done with a reaction with proline or another nitrogen-containing heterocycle, which control chirality with that of the catalyst. This has been theorized to be caused by the restriction of undesired (E)-isomer by preventing the ketone from accessing non-reactive tautomers. Generally, a Mannich reaction is the combination of an amine, a ketone with a β-acidic proton and aldehyde to create a condensed product in a β-addition to the ketone. This occurs through an attack on the ketone with a suitable catalytic-amine unto its electron-starved carbon, from which an imine is created. This then undergoes electrophilic addition with a compound containing an acidic proton. It is theoretically possible for either of the carbonyl-containing molecules to create diastereomers, but with the addition of catalysts which restrict addition as of the enamine creation, it is possible to extract a single product with limited purification steps and in some cases as reported by List et al.; practical one-pot syntheses are possible. The process of selecting a carbonyl-group gives the reaction a direct versus indirect distinction, wherein the latter case represents pre-formed products restricting the reaction's pathway and the other does not. Ketimines selects a reaction group, and circumvent a requirement for indirect pathways.