Related Research Articles

Fabry disease, also known as Anderson–Fabry disease, is a rare genetic disease that can affect many parts of the body, including the kidneys, heart, and skin. Fabry disease is one of a group of conditions known as lysosomal storage diseases. The genetic mutation that causes Fabry disease interferes with the function of an enzyme that processes biomolecules known as sphingolipids, leading to these substances building up in the walls of blood vessels and other organs. It is inherited in an X-linked manner.

Alpha-mannosidosis is a lysosomal storage disorder, first described by Swedish physician Okerman in 1967. In humans it is known to be caused by an autosomal recessive genetic mutation in the gene MAN2B1, located on chromosome 19, affecting the production of the enzyme alpha-D-mannosidase, resulting in its deficiency. Consequently, if both parents are carriers, there will be a 25% chance with each pregnancy that the defective gene from both parents will be inherited, and the child will develop the disease. There is a two in three chance that unaffected siblings will be carriers. In livestock alpha-mannosidosis is caused by chronic poisoning with swainsonine from locoweed.
Iduronidase, sold as Aldurazyme, is an enzyme with the systematic name glycosaminoglycan α-L-iduronohydrolase. It catalyses the hydrolysis of unsulfated α-L-iduronosidic linkages in dermatan sulfate.
Alglucosidase alfa, sold under the brand name Myozyme among others, is an enzyme replacement therapy (ERT) orphan drug for treatment of Pompe disease, a rare lysosomal storage disorder (LSD). Chemically, the drug is an analog of the enzyme that is deficient in patients affected by Pompe disease, alpha-glucosidase. It is the first drug available to treat this disease.

Beta-mannosidosis, also called lysosomal beta-mannosidase deficiency, is a disorder of oligosaccharide metabolism caused by decreased activity of the enzyme beta-mannosidase. This enzyme is coded for by the gene MANBA, located at 4q22-25. Beta-mannosidosis is inherited in an autosomal recessive manner. Affected individuals appear normal at birth, and can have a variable clinical presentation. Infantile onset forms show severe neurodegeneration, while some children have intellectual disability. Hearing loss and angiokeratomas are common features of the disease.
Dupilumab, sold under the brand name Dupixent, is a monoclonal antibody blocking interleukin 4 and interleukin 13, used for allergic diseases such as eczema, asthma and nasal polyps which result in chronic sinusitis. It is also used for the treatment of eosinophilic esophagitis and prurigo nodularis.
Eptinezumab, sold under the brand name Vyepti, is a medication used for the preventive treatment of migraine in adults. It is a monoclonal antibody that targets calcitonin gene-related peptides (CGRP) alpha and beta. It is administered by intravenous infusion.
Lanadelumab, sold under the brand name Takhzyro, is a human monoclonal antibody that targets plasma kallikrein (pKal) in order to promote prevention of angioedema in people with hereditary angioedema. Lanadelumab, was approved in the United States as the first monoclonal antibody indicated for prophylactic treatment to prevent hereditary angioedema attacks. Lanadelumab is the first treatment for hereditary angioedema prevention made by using cells within a lab, not human plasma.
Vestronidase alfa, sold under brand name Mepsevii, is a drug for the treatment of Sly syndrome. It is a recombinant form of the human enzyme beta-glucuronidase. It was approved in the United States in November 2017, to treat children and adults with an inherited metabolic condition called mucopolysaccharidosis type VII, also known as Sly syndrome. MPS VII is an extremely rare, progressive condition that affects most tissues and organs.

Zuranolone, sold under the brand name Zurzuvae, is a medication used for the treatment of postpartum depression. It is taken by mouth.
Lecanemab, sold under the brand name Leqembi, is a monoclonal antibody medication used for the treatment of Alzheimer's disease. Lecanemab is an amyloid beta-directed antibody. It is given via intravenous infusion. The most common side effects of lecanemab include headache, infusion-related reactions and amyloid-related imaging abnormalities, a side effect known to occur with the class of antibodies targeting amyloid.
Satralizumab, sold under the brand name Enspryng, is a humanized monoclonal antibody medication that is used for the treatment of neuromyelitis optica spectrum disorder (NMOSD), a rare autoimmune disease. The drug is being developed by Chugai Pharmaceutical, a subsidiary of Roche.
Sutimlimab, sold under the brand name Enjaymo, is a monoclonal antibody that is used to treat adults with cold agglutinin disease (CAD). It is given by intravenous infusion. Sutimlimab prevents complement-enhanced activation of autoimmune human B cells in vitro.
Lumasiran, sold under the brand name Oxlumo, is a medication for the treatment of primary hyperoxaluria type 1 (PH1).
Avalglucosidase alfa, sold under the brand name Nexviazyme, is an enzyme replacement therapy medication used for the treatment of glycogen storage disease type II.
Efgartigimod alfa, sold under the brand name Vyvgart, is a medication used to treat myasthenia gravis. Efgartigimod alfa is a neonatal Fc receptor blocker and is a new class of medication. It is an antibody fragment that binds to the neonatal Fc receptor (FcRn), preventing FcRn from recycling immunoglobulin G (IgG) back into the blood. The medication causes a reduction in overall levels of IgG, including the abnormal acetylcholine receptor (AChR) antibodies that are present in myasthenia gravis. It is also available coformulated with hyaluronidase.
Olipudase alfa, sold under the brand name Xenpozyme, is a medication used for the treatment of non-central nervous system (CNS) manifestations of acid sphingomyelinase deficiency (ASMD) type A/B or type B.
Pegunigalsidase alfa, sold under the brand name Elfabrio, is an enzyme replacement therapy for the treatment of Fabry disease. It is a recombinant human α-galactosidase-A. It is a hydrolytic lysosomal neutral glycosphingolipid-specific enzyme.
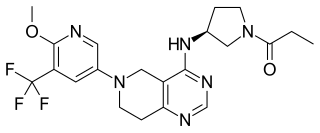
Leniolisib (INN), sold under the brand name Joenja, is a medication used for the treatment of activated phosphoinositide 3-kinase delta syndrome (APDS). Leniolisib is a kinase inhibitor.
Pozelimab, sold under the brand name Veopoz, is a recombinant monoclonal antibody used for the treatment of CD55-deficient protein-losing enteropathy, also known as CHAPLE disease. Pozelimab is a complement inhibitor. It is produced using recombinant DNA technology in Chinese hamster ovary cells.
References
- ↑ "Lamzede (velmanase alfa-tycv) for injection, for intravenous use" (PDF). Archived (PDF) from the original on 22 February 2023. Retrieved 21 February 2023.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 "FDA approves first enzyme replacement therapy for rare alpha-mannosidosis". U.S. Food and Drug Administration (FDA). 17 February 2023. Retrieved 17 February 2023.
 This article incorporates text from this source, which is in the public domain .
This article incorporates text from this source, which is in the public domain . - 1 2 3 "Lamzede EPAR". European Medicines Agency (EMA). 17 September 2018. Archived from the original on 11 August 2020. Retrieved 25 August 2020.
- ↑ Malm D, Nilssen Ø (July 2019) [October 2001]. "Alpha-Mannosidosis". In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Gripp KW, Mirzaa GM, Amemiya A (eds.). GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle. PMID 20301570. Archived from the original on 26 October 2020. Retrieved 29 May 2022.
{{cite book}}: CS1 maint: location missing publisher (link) - 1 2 "Chiesi Group receives the European Marketing Authorisation for Lamzede" (Press release). 4 April 2018. Archived from the original on 19 April 2021. Retrieved 24 August 2020.
- ↑ "Chiesi Global Rare Diseases Announces FDA Approval of Lamzede (velmanase alfa-tycv) for Alpha-Mannosidosis" (Press release). Chiesi Global Rare Diseases. 16 February 2023. Archived from the original on 17 February 2023. Retrieved 17 February 2023– via PR Newswire.
- ↑ World Health Organization (2016). "International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 75". WHO Drug Information. 30 (1). hdl: 10665/331046 .