Related Research Articles
An allele is a variant of the sequence of nucleotides at a particular location, or locus, on a DNA molecule.

A genetic disorder is a health problem caused by one or more abnormalities in the genome. It can be caused by a mutation in a single gene (monogenic) or multiple genes (polygenic) or by a chromosome abnormality. Although polygenic disorders are the most common, the term is mostly used when discussing disorders with a single genetic cause, either in a gene or chromosome. The mutation responsible can occur spontaneously before embryonic development, or it can be inherited from two parents who are carriers of a faulty gene or from a parent with the disorder. When the genetic disorder is inherited from one or both parents, it is also classified as a hereditary disease. Some disorders are caused by a mutation on the X chromosome and have X-linked inheritance. Very few disorders are inherited on the Y chromosome or mitochondrial DNA.
Online Mendelian Inheritance in Man (OMIM) is a continuously updated catalog of human genes and genetic disorders and traits, with a particular focus on the gene-phenotype relationship. As of 28 June 2019, approximately 9,000 of the over 25,000 entries in OMIM represented phenotypes; the rest represented genes, many of which were related to known phenotypes.
Diamond–Blackfan anemia (DBA) is a congenital erythroid aplasia that usually presents in infancy. DBA causes low red blood cell counts (anemia), without substantially affecting the other blood components, which are usually normal. This is in contrast to Shwachman–Bodian–Diamond syndrome, in which the bone marrow defect results primarily in neutropenia, and Fanconi anemia, where all cell lines are affected resulting in pancytopenia. There is a risk to develop acute myelogenous leukemia (AML) and certain other cancers.

22q13 deletion syndrome, known as Phelan–McDermid syndrome (PMS), is a genetic disorder caused by deletions or rearrangements on the q terminal end of chromosome 22. Any abnormal genetic variation in the q13 region that presents with significant manifestations (phenotype) typical of a terminal deletion may be diagnosed as 22q13 deletion syndrome. There is disagreement among researchers as to the exact definition of 22q13 deletion syndrome. The Developmental Synaptopathies Consortium defines PMS as being caused by SHANK3 mutations, a definition that appears to exclude terminal deletions. The requirement to include SHANK3 in the definition is supported by many but not by those who first described 22q13 deletion syndrome.
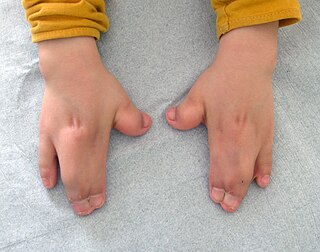
Acrocephalosyndactyly is a group of congenital conditions characterized by irregular features of the face and skull (craniosynostosis) and hands and feet (syndactyly). Craniosynostosis occurs when the cranial sutures, the fibrous tissue connecting the skull bones, fuse the cranial bones early in development. Cranial sutures allow the skull bones to continue growing until they fuse at age 24. Premature fusing of the cranial sutures can result in alterations to the skull shape and interfere with brain growth. Syndactyly occurs when digits of the hands or feet are fused together. When polydactyly is also present, the classification is acrocephalopolysyndactyly. Polydactyly occurs when the hands or feet possess additional digits. Acrocephalosyndactyly is usually diagnosed after birth, although prenatal diagnosis is sometimes possible if the genetic variation is present in family members, as the conditions are typically inherited in an autosomal dominant pattern Treatment often involves surgery in early childhood to correct for craniosynostosis and syndactyly.

Lujan–Fryns syndrome (LFS) is an X-linked genetic disorder that causes mild to moderate intellectual disability and features described as Marfanoid habitus, referring to a group of physical characteristics similar to those found in Marfan syndrome. These features include a tall, thin stature and long, slender limbs. LFS is also associated with psychopathology and behavioral abnormalities, and it exhibits a number of malformations affecting the brain and heart. The disorder is inherited in an X-linked dominant manner, and is attributed to a missense mutation in the MED12 gene. There is currently no treatment or therapy for the underlying MED12 malfunction, and the exact cause of the disorder remains unclear.

Aravinda Chakravarti is a human geneticist and expert in computational biology, and Director of the Center For Human Genetics & Genomics at New York University. He was the 2008 President of the American Society of Human Genetics. Chakravarti became a co-Editor-in-Chief of the journal Genome Research in 1995, and of the Annual Review of Genomics and Human Genetics' in 2005.

Pontocerebellar hypoplasia (PCH) is a heterogeneous group of rare neurodegenerative disorders caused by genetic mutations and characterised by progressive atrophy of various parts of the brain such as the cerebellum or brainstem. Where known, these disorders are inherited in an autosomal recessive fashion. There is no known cure for PCH.

Mental retardation and microcephaly with pontine and cerebellar hypoplasia (MICPCH) – also known as mental retardation, X-linked, syndromic, Najm type (MRXSNA); X-linked intellectual deficit, Najm type; intellectual developmental disorder, X-linked, syndromic, Najm type; X-linked intellectual disability–microcephaly–pontocerebellar hypoplasia syndrome; and by variations of these terms – is a rare X-linked dominant genetic disorder of infants characterised by intellectual disability and pontocerebellar hypoplasia. It usually affects females; many males die before birth or not long after.
In bioinformatics, a Gene Disease Database is a systematized collection of data, typically structured to model aspects of reality, in a way to comprehend the underlying mechanisms of complex diseases, by understanding multiple composite interactions between phenotype-genotype relationships and gene-disease mechanisms. Gene Disease Databases integrate human gene-disease associations from various expert curated databases and text mining derived associations including Mendelian, complex and environmental diseases.
The Monarch Initiative is a large scale bioinformatics web resource focused on leveraging existing biomedical knowledge to connect genotypes with phenotypes in an effort to aid research that combats genetic diseases. Monarch does this by integrating multi-species genotype, phenotype, genetic variant and disease knowledge from various existing biomedical data resources into a centralized and structured database. While this integration process has been traditionally done manually by basic researchers and clinicians on a case-by-case basis, The Monarch Initiative provides an aggregated and structured collection of data and tools that make biomedical knowledge exploration more efficient and effective.
ZC4H2 is a protein-coding gene located on the X-chromosome. This gene encodes a protein which is a member of the so-called zinc finger domain-containing protein family. There is currently very limited understanding about the ZC4H2 gene and its protein function.
GeneMatcher is an online service and database that aims to match clinicians studying patients with a rare disease presentation based on genes of interest. When two or more clinicians submit the same gene to the database, the service matches them together to allow them to compare cases. It also allows matching genes from animal models to human cases. The service aims to establish novel relationships between genes and genetic diseases of unknown cause.

Jared C. Roach is an American biologist who invented the pairwise end sequencing strategy while a graduate student at the University of Washington.

Lowry-Wood syndrome, also simply known as LWS, is a very rare genetic disorder which is characterized by dysplasia of the epiphysis, low height/short stature, microcephaly, developmental delay, intellectual disabilities, and congenital nystagmus. Less common features include coxa vara and retinitis pigmentosa. Only 10 cases of this disorder have been described in medical literature. This disorder is associated with mutations in the RNU4ATAC gene, on chromosome 2q14.2
Chudley–Mccullough syndrome is a rare genetic disorder which is characterized by bilateral congenital hearing loss associated with brain malformations. It is a type of syndromic deafness.
Meacham syndrome is a rare genetic disorder which is characterized by lung, diaphragmatic and genitourinary anomalies.

Czech dysplasia metatarsal type is a rare type of Czech dysplasia which is characterized primarily by bone anomalies.
References
- 1 2 3 4 Genetics, American College of Medical (2021-04-13). "Geneticist and Pediatrician Dr. Ada Hamosh Receives David L. Rimoin Lifetime Achievement Award in Medical Genetics from the ACMG Foundation for Genetic and Genomic Medicine". PR Newswire. Retrieved 2024-11-14.
- ↑ Rasmussen, Sonja A.; Hamosh, Ada (2021). "Memories of Victor A. McKusick". American Journal of Medical Genetics Part A. 185 (11): 3377–3383. doi:10.1002/ajmg.a.62431. ISSN 1552-4825.
- ↑ Hamosh, Ada; Amberger, Joanna S.; Bocchini, Carol; Scott, Alan F.; Rasmussen, Sonja A. (2021). "Online Mendelian Inheritance in Man ( OMIM ®): Victor McKusick 's magnum opus". American Journal of Medical Genetics Part A. 185 (11): 3259–3265. doi: 10.1002/ajmg.a.62407 . ISSN 1552-4825. PMC 8596664 . PMID 34169650.
- ↑ Amberger, Joanna S.; Bocchini, Carol A.; Scott, Alan F.; Hamosh, Ada (2019-01-08). "OMIM.org: leveraging knowledge across phenotype-gene relationships". Nucleic Acids Research. 47 (D1): D1038 –D1043. doi: 10.1093/nar/gky1151 . ISSN 1362-4962. PMC 6323937 . PMID 30445645.
- ↑ Amberger, Joanna S.; Hamosh, Ada (2017-06-27). "Searching Online Mendelian Inheritance in Man (OMIM): A Knowledgebase of Human Genes and Genetic Phenotypes". Current Protocols in Bioinformatics. 58: 1.2.1–1.2.12. doi: 10.1002/cpbi.27 . ISSN 1934-340X. PMC 5662200 . PMID 28654725.
- ↑ Baxter, Samantha M. (2022). "Centers for Mendelian Genomics: A decade of facilitating gene discovery". Genetics in Medicine. 24 (4): 784–797. doi: 10.1016/j.gim.2021.12.005 . PMC 9119004 . PMID 35148959.
- ↑ Wohler, Elizabeth; Martin, Renan; Griffith, Sean (2021). "PhenoDB, GeneMatcher and VariantMatcher, tools for analysis and sharing of sequence data". Orphanet Journal of Rare Diseases. 16 (1): 365. doi: 10.1186/s13023-021-01916-z . ISSN 1750-1172. PMC 8371856 . PMID 34407837.
- ↑ Morrissey, Laurie (NIH/NCI) [C] (2015-03-23). "April 1: Dr. Ada Hamosh and Dr. Nara Sobreira, PhenoDB: A Tool for Collection and Analysis of Phenotypic and Genomic Data". NCI Wiki. Retrieved 2024-11-14.
- ↑ Sobreira, Nara; Schiettecatte, François; Valle, David; Hamosh, Ada (2015). "GeneMatcher: a matching tool for connecting investigators with an interest in the same gene". Human Mutation. 36 (10): 928–930. doi: 10.1002/humu.22844 . ISSN 1098-1004. PMC 4833888 . PMID 26220891.
- ↑ Dyke, Stephanie O. M.; Knoppers, Bartha M.; Hamosh, Ada (2017). ""Matching" consent to purpose: The example of the Matchmaker Exchange". Human Mutation. 38 (10): 1281–1285. doi: 10.1002/humu.23278 . ISSN 1098-1004. PMC 5669800 . PMID 28699299.
- ↑ Lee-Barber, Jasmine; Kulo, Violet; Lehmann, Harold; Hamosh, Ada; Bodurtha, Joann (2019). "Bioinformatics for medical students: a 5-year experience using OMIM® in medical student education". Genetics in Medicine. 21 (2): 493–497. doi: 10.1038/s41436-018-0076-7 . ISSN 1530-0366. PMID 29930391.
- ↑ Diehl, Adam C.; Reader, Lauren; Hamosh, Ada; Bodurtha, Joann N. (2015). "Horizontal integration of OMIM across the medical school preclinical curriculum for early reinforcement of clinical genetics principles". Genetics in Medicine. 17 (2). Elsevier BV: 158–163. doi: 10.1038/gim.2014.84 . ISSN 1098-3600. PMC 4859213 . PMID 25032988.
- ↑ de Macena Sobreira, Nara Lygia; Hamosh, Ada (2021). "Next-generation sequencing and the evolution of data sharing". American Journal of Medical Genetics Part A. 185 (9): 2633–2635. doi:10.1002/ajmg.a.62239. ISSN 1552-4825.
- ↑ International, HUGO (2024-07-15). "HUGO President & Executive Board Members". HUGO International. Retrieved 2024-11-14.
- ↑ "ASHG 2024: Stalwarts of Human Genetics Receive Professional Awards". GEN - Genetic Engineering and Biotechnology News. 2024-11-05. Retrieved 2024-11-14.
- ↑ "NORD Announces 2023 Rare Impact Award Honorees and 40th Anniversary Celebration". National Organization for Rare Disorders. 2023-02-27. Retrieved 2024-11-14.
| International | |
|---|---|
| National | |
| Academics | |
| Other | |