Related Research Articles
Sharpless asymmetric dihydroxylation is the chemical reaction of an alkene with osmium tetroxide in the presence of a chiral quinine ligand to form a vicinal diol. The reaction has been applied to alkenes of virtually every substitution, often high enantioselectivities are realized, with the chiral outcome controlled by the choice of dihydroquinidine (DHQD) vs dihydroquinine (DHQ) as the ligand. Asymmetric dihydroxylation reactions are also highly site selective, providing products derived from reaction of the most electron-rich double bond in the substrate.
In chemistry, an electrophile is a chemical species that forms bonds with nucleophiles by accepting an electron pair. Because electrophiles accept electrons, they are Lewis acids. Most electrophiles are positively charged, have an atom that carries a partial positive charge, or have an atom that does not have an octet of electrons.
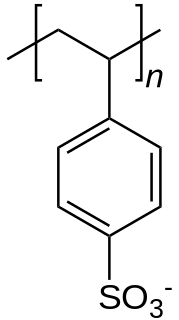
In chemistry, a counterion is the ion that accompanies an ionic species in order to maintain electric neutrality. In table salt the sodium ion is the counterion for the chloride ion and vice versa.
In chemistry, a phase-transfer catalyst or PTC is a catalyst that facilitates the transition of a reactant from one phase into another phase where reaction occurs. Phase-transfer catalysis is a special form of heterogeneous catalysis. Ionic reactants are often soluble in an aqueous phase but insoluble in an organic phase in the absence of the phase-transfer catalyst. The catalyst functions like a detergent for solubilizing the salts into the organic phase. Phase-transfer catalysis refers to the acceleration of the reaction upon the addition of the phase-transfer catalyst.
Dihydroxylation is the process by which an alkene is converted into a vicinal diol. Although there are many routes to accomplish this oxidation, the most common and direct processes use a high-oxidation-state transition metal. The metal is often used as a catalyst, with some other stoichiometric oxidant present. In addition, other transition metals and non-transition metal methods have been developed and used to catalyze the reaction.

In organic chemistry, organocatalysis is a form of catalysis in which the rate of a chemical reaction is increased by an organic catalyst. This "organocatalyst" consists of carbon, hydrogen, sulfur and other nonmetal elements found in organic compounds. Because of their similarity in composition and description, they are often mistaken as a misnomer for enzymes due to their comparable effects on reaction rates and forms of catalysis involved.
In chemistry, bis(oxazoline) ligands (often abbreviated BOX ligands) are a class of privileged chiral ligands containing two oxazoline rings. They are typically C2‑symmetric and exist in a wide variety of forms; with structures based around CH2 or pyridine linkers being particularly common (often generalised BOX and PyBOX respectively). The coordination complexes of bis(oxazoline) ligands are used in asymmetric catalysis. These ligands are examples of C2-symmetric ligands.
Asymmetric hydrogenation is a chemical reaction that adds two atoms of hydrogen to a target (substrate) molecule with three-dimensional spatial selectivity. Critically, this selectivity does not come from the target molecule itself, but from other reagents or catalysts present in the reaction. This allows spatial information to transfer from one molecule to the target, forming the product as a single enantiomer. The chiral information is most commonly contained in a catalyst and, in this case, the information in a single molecule of catalyst may be transferred to many substrate molecules, amplifying the amount of chiral information present. Similar processes occur in nature, where a chiral molecule like an enzyme can catalyse the introduction of a chiral centre to give a product as a single enantiomer, such as amino acids, that a cell needs to function. By imitating this process, chemists can generate many novel synthetic molecules that interact with biological systems in specific ways, leading to new pharmaceutical agents and agrochemicals. The importance of asymmetric hydrogenation in both academia and industry contributed to two of its pioneers — William Standish Knowles and Ryōji Noyori — being awarded one half of the 2001 Nobel Prize in Chemistry.

DuPhos is a class of organophosphorus compound that are used ligands for asymmetric synthesis. The name DuPhos is derived from (1) the chemical company that sponsored the research leading to this ligand's invention, DuPont and (2) the compound is a diphosphine ligand type. Specifically it is classified as a C2-symmetric ligand, consisting of two phospholanes rings affixed to a benzene ring.
Organogold chemistry is the study of compounds containing gold–carbon bonds. They are studied in academic research, but have not received widespread use otherwise. The dominant oxidation states for organogold compounds are I with coordination number 2 and a linear molecular geometry and III with CN = 4 and a square planar molecular geometry. The first organogold compound discovered was gold(I) carbide Au2C2, which was first prepared in 1900.
In Lewis acid catalysis of organic reactions, a metal-based Lewis acid acts as an electron pair acceptor to increase the reactivity of a substrate. Common Lewis acid catalysts are based on main group metals such as aluminum, boron, silicon, and tin, as well as many early and late d-block metals. The metal atom forms an adduct with a lone-pair bearing electronegative atom in the substrate, such as oxygen, nitrogen, sulfur, and halogens. The complexation has partial charge-transfer character and makes the lone-pair donor effectively more electronegative, activating the substrate toward nucleophilic attack, heterolytic bond cleavage, or cycloaddition with 1,3-dienes and 1,3-dipoles.

Hydrogen-bond catalysis is a type of organocatalysis that relies on use of hydrogen bonding interactions to accelerate and control organic reactions. In biological systems, hydrogen bonding plays a key role in many enzymatic reactions, both in orienting the substrate molecules and lowering barriers to reaction. However, chemists have only recently attempted to harness the power of using hydrogen bonds to perform catalysis, and the field is relatively undeveloped compared to research in Lewis acid catalysis.

Phosphinooxazolines are a class of chiral ligands used in asymmetric catalysis. Their complexes are particularly effective at generating single enatiomers in reactions involving highly symmetric transition states, such as allylic substitutions, which are typically difficult to perform stereoselectively. The ligands are bidentate and have been shown to be hemilabile with the softer P‑donor being more firmly bound than the harder N‑donor.
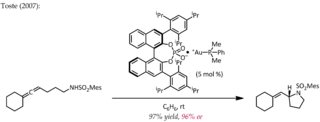
Asymmetric counteranion directed catalysis (ACDC) or chiral anion catalysis in enantioselective synthesis is the "induction of enantioselectivity in a reaction proceeding through a cationic intermediate by means of ion pairing with a chiral, enantiomerically pure anion provided by the catalyst". Although chiral Brønsted acid catalyzed reactions may well fall into this category of catalysis under the definition given here, the extent of proton transfer and the demarcation between hydrogen bonding and full proton transfer is often ambiguous. Hence, some authors may exclude ion pair formation by proton transfer as a type of chiral counteranion catalysis. The discussion below will focus on chiral ion pairs generated through means other than proton transfer.
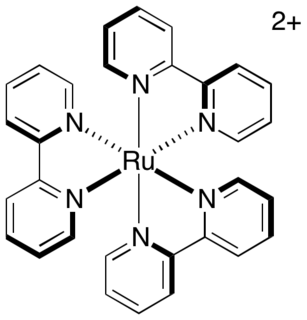
Photoredox catalysis is a branch of photochemistry that uses single-electron transfer. Photoredox catalysts are generally drawn from three classes of materials: transition-metal complexes, organic dyes, and semiconductors. While organic photoredox catalysts were dominant throughout the 1990s and early 2000s, soluble transition-metal complexes are more commonly used today.
Cycloisomerization is any isomerization in which the cyclic isomer of the substrate is produced in the reaction coordinate. The greatest advantage of cycloisomerization reactions is its atom economical nature, by design nothing is wasted, as every atom in the starting material is present in the product. In most cases these reactions are mediated by a transition metal catalyst, in few cases organocatalysts and rarely do they occur under thermal conditions. These cyclizations are able to be performed with excellent levels of selectivity in numerous cases and have transformed cycloisomerization into a powerful tool for unique and complex molecular construction. Cycloisomerization is a very broad topic in organic synthesis and many reactions that would be categorized as such exist. Two basic classes of these reactions are intramolecular Michael addition and Intramolecular Diels–Alder reactions. Under the umbrella of cycloisomerization, enyne and related olefin cycloisomerizations are the most widely used and studied reactions.
Cobalt(II)–porphyrin catalysis is a process in which a Co(II) porphyrin complex acts as a catalyst, inducing and accelerating a chemical reaction.
In homogeneous catalysis, C2-symmetric ligands refer to ligands that lack mirror symmetry but have C2 symmetry. Such ligands are usually bidentate and are valuable in catalysis. The C2 symmetry of ligands limits the number of possible reaction pathways and thereby increases enantioselectivity, relative to asymmetrical analogues. C2-symmetric ligands are a subset of chiral ligands. Chiral ligands, including C2-symmetric ligands, combine with metals or other groups to form chiral catalysts. These catalysts engage in enantioselective chemical synthesis, in which chirality in the catalyst yields chirality in the reaction product.
F. Dean Toste is the Gerald E. K. Branch Distinguished Professor of Chemistry at the University of California, Berkeley and Faculty Scientist at the Chemical Sciences Division of Lawrence Berkeley National Lab. He is a prominent figure in the field of organic chemistry and is best known for his contributions to gold chemistry and asymmetric ion-pairing catalysis. Toste was elected a member of the National Academy of Sciences in 2020, and a member of the American Academy of Arts and Sciences in 2018.

The nitro-Mannich reaction is the nucleophilic addition of a nitroalkane to an imine, resulting in the formation of a beta-nitroamine. With the reaction involving the addition of an acidic carbon nucleophile to a carbon-heteroatom double bond, the nitro-Mannich reaction is related to some of the most fundamental carbon-carbon bond forming reactions in organic chemistry, including the aldol reaction, Henry reaction and Mannich reaction.
References
- ↑ Brak, K. and Jacobsen, E. N. (2013), Asymmetric Ion-Pairing Catalysis. Angew. Chem. Int. Ed., 52: 534–561. doi : 10.1002/anie.201205449
- ↑ Efficient catalytic asymmetric alkylations. 1. Enantioselective synthesis of (+)-indacrinone via chiral phase-transfer catalysis Ulf H. Dolling, Paul Davis, and Edward J. J. Grabowski Journal of the American Chemical Society 1984 106 (2), 446-447 doi : 10.1021/ja00314a045
- ↑ Chiral crown complexes catalyse Michael addition reactions to give adducts in high optical yields Donald J. Cram and G. D. Y. Sogah J. Chem. Soc., Chem. Commun., 1981, 625-628 doi : 10.1039/C39810000625
- ↑ A Novel Example of Chiral Counteranion Induced Enantioselective Metal Catalysis: The Importance of Ion-Pairing in Copper-Catalyzed Olefin Aziridination and Cyclopropanation David B. Llewellyn,Dan Adamson, and, and Bruce A. Arndtsen Organic Letters 2000 2 (26), 4165-4168 doi : 10.1021/ol000303y
- ↑ Chiral Anion-Mediated Asymmetric Ring Opening of meso-Aziridinium and Episulfonium Ions Gregory L. Hamilton, Toshio Kanai, and F. Dean Toste Journal of the American Chemical Society 2008 130 (45), 14984-14986 doi : 10.1021/ja806431d