Proteins are large biomolecules and macromolecules that comprise one or more long chains of amino acid residues. Proteins perform a vast array of functions within organisms, including catalysing metabolic reactions, DNA replication, responding to stimuli, providing structure to cells and organisms, and transporting molecules from one location to another. Proteins differ from one another primarily in their sequence of amino acids, which is dictated by the nucleotide sequence of their genes, and which usually results in protein folding into a specific 3D structure that determines its activity.

Dihydrofolate reductase, or DHFR, is an enzyme that reduces dihydrofolic acid to tetrahydrofolic acid, using NADPH as an electron donor, which can be converted to the kinds of tetrahydrofolate cofactors used in 1-carbon transfer chemistry. In humans, the DHFR enzyme is encoded by the DHFR gene. It is found in the q14.1 region of chromosome 5.
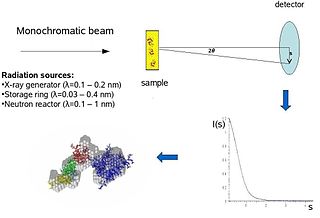
Biological small-angle scattering is a small-angle scattering method for structure analysis of biological materials. Small-angle scattering is used to study the structure of a variety of objects such as solutions of biological macromolecules, nanocomposites, alloys, and synthetic polymers. Small-angle X-ray scattering (SAXS) and small-angle neutron scattering (SANS) are the two complementary techniques known jointly as small-angle scattering (SAS). SAS is an analogous method to X-ray and neutron diffraction, wide angle X-ray scattering, as well as to static light scattering. In contrast to other X-ray and neutron scattering methods, SAS yields information on the sizes and shapes of both crystalline and non-crystalline particles. When used to study biological materials, which are very often in aqueous solution, the scattering pattern is orientation averaged.

Helix-turn-helix is a DNA-binding protein (DBP). The helix-turn-helix (HTH) is a major structural motif capable of binding DNA. Each monomer incorporates two α helices, joined by a short strand of amino acids, that bind to the major groove of DNA. The HTH motif occurs in many proteins that regulate gene expression. It should not be confused with the helix–loop–helix motif.

Sir Alan Roy Fersht is a British chemist at the MRC Laboratory of Molecular Biology, Cambridge, and an Emeritus Professor in the Department of Chemistry at the University of Cambridge. He was Master of Gonville and Caius College, Cambridge from 2012 to 2018. He works on protein folding, and is sometimes described as a founder of protein engineering.
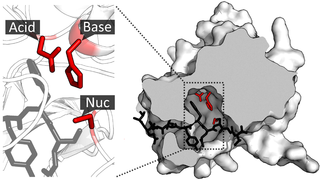
A catalytic triad is a set of three coordinated amino acids that can be found in the active site of some enzymes. Catalytic triads are most commonly found in hydrolase and transferase enzymes. An acid-base-nucleophile triad is a common motif for generating a nucleophilic residue for covalent catalysis. The residues form a charge-relay network to polarise and activate the nucleophile, which attacks the substrate, forming a covalent intermediate which is then hydrolysed to release the product and regenerate free enzyme. The nucleophile is most commonly a serine or cysteine amino acid, but occasionally threonine or even selenocysteine. The 3D structure of the enzyme brings together the triad residues in a precise orientation, even though they may be far apart in the sequence.

In molecular biology, an intrinsically disordered protein (IDP) is a protein that lacks a fixed or ordered three-dimensional structure, typically in the absence of its macromolecular interaction partners, such as other proteins or RNA. IDPs range from fully unstructured to partially structured and include random coil, molten globule-like aggregates, or flexible linkers in large multi-domain proteins. They are sometimes considered as a separate class of proteins along with globular, fibrous and membrane proteins.
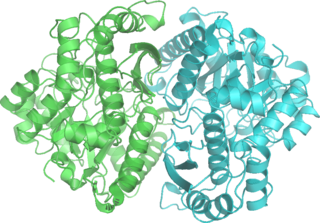
Phosphopyruvate hydratase, usually known as enolase, is a metalloenzyme (EC 4.2.1.11) that catalyses the conversion of 2-phosphoglycerate (2-PG) to phosphoenolpyruvate (PEP), the ninth and penultimate step of glycolysis. The chemical reaction is:
Hans Charles Freeman AM, FAA was a German-born Australian bioinorganic chemist, protein crystallographer, and professor of inorganic chemistry who spent most of his academic career at the University of Sydney. His best known contributions to chemistry were his work explaining the unusual structural, electrochemical, and spectroscopic properties of blue copper proteins, particularly plastocyanin. He also introduced protein crystallography to Australia and was a strong advocate for courses to ensure Australian scientists have good access to "big science" facilities. Freeman has received numerous honours, including being elected a Fellow of the Australian Academy of Science (FAA) and appointed a Member of the Order of Australia (AM) by the Australian Government. He was a charismatic lecturer who voluntarily continued teaching well into his formal retirement and imbued his students with a love of science.
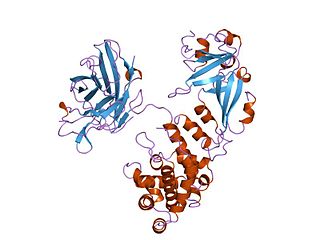
Diphtheria toxin is an exotoxin secreted mainly by Corynebacterium diphtheriae but also by Corynebacterium ulcerans and Corynebacterium pseudotuberculosis, the pathogenic bacterium that causes diphtheria. The toxin gene is encoded by a prophage called corynephage β. The toxin causes the disease in humans by gaining entry into the cell cytoplasm and inhibiting protein synthesis.
Phi value analysis, analysis, or -value analysis is an experimental protein engineering technique for studying the structure of the folding transition state of small protein domains that fold in a two-state manner. The structure of the folding transition state is hard to find using methods such as protein NMR or X-ray crystallography because folding transitions states are mobile and partly unstructured by definition. In -value analysis, the folding kinetics and conformational folding stability of the wild-type protein are compared with those of point mutants to find phi values. These measure the mutant residue's energetic contribution to the folding transition state, which reveals the degree of native structure around the mutated residue in the transition state, by accounting for the relative free energies of the unfolded state, the folded state, and the transition state for the wild-type and mutant proteins.

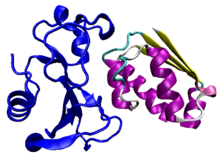
Barnase (a portmanteau of "BActerial" "RiboNucleASE") is a bacterial protein that consists of 110 amino acids and has ribonuclease activity. It is synthesized and secreted by the bacterium Bacillus amyloliquefaciens, but is lethal to the cell when expressed without its inhibitor barstar. The inhibitor binds to and occludes the ribonuclease active site, preventing barnase from damaging the cell's RNA after it has been synthesized but before it has been secreted. The barnase/barstar complex is noted for its extraordinarily tight protein-protein binding, with an on-rate of 108s−1M−1.

Peptidyl-prolyl cis-trans isomerase FKBP1A is an enzyme that in humans is encoded by the FKBP1A gene. It is also commonly referred to as FKBP-12 or FKBP12 and is a member of a family of FK506-binding proteins (FKBPs).

Calmodulin 3 is a protein that in humans is encoded by the CALM3 gene.
Tyrosine—tRNA ligase, also known as tyrosyl-tRNA synthetase is an enzyme that is encoded by the gene YARS. Tyrosine—tRNA ligase catalyzes the chemical reaction

U1 small nuclear ribonucleoprotein A is a protein that in humans is encoded by the SNRPA gene.

Morpheeins are proteins that can form two or more different homo-oligomers, but must come apart and change shape to convert between forms. The alternate shape may reassemble to a different oligomer. The shape of the subunit dictates which oligomer is formed. Each oligomer has a finite number of subunits (stoichiometry). Morpheeins can interconvert between forms under physiological conditions and can exist as an equilibrium of different oligomers. These oligomers are physiologically relevant and are not misfolded protein; this distinguishes morpheeins from prions and amyloid. The different oligomers have distinct functionality. Interconversion of morpheein forms can be a structural basis for allosteric regulation, an idea noted many years ago, and later revived. A mutation that shifts the normal equilibrium of morpheein forms can serve as the basis for a conformational disease. Features of morpheeins can be exploited for drug discovery. The dice image represents a morpheein equilibrium containing two different monomeric shapes that dictate assembly to a tetramer or a pentamer. The one protein that is established to function as a morpheein is porphobilinogen synthase, though there are suggestions throughout the literature that other proteins may function as morpheeins.
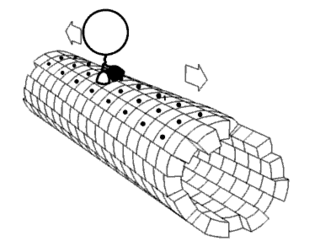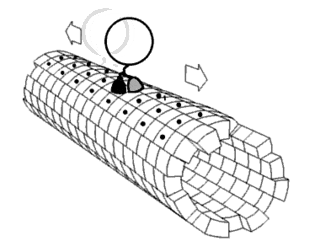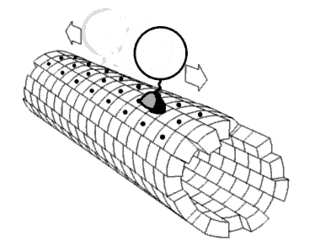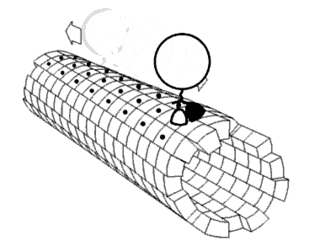
Proteins are generally thought to adopt unique structures determined by their amino acid sequences. However, proteins are not strictly static objects, but rather populate ensembles of conformations. Transitions between these states occur on a variety of length scales and time scales , and have been linked to functionally relevant phenomena such as allosteric signaling and enzyme catalysis.
Andreas Matouschek is a biochemist at The University of Texas at Austin, where he is a professor in the College of Natural Sciences. His graduate work with Alan Fersht resulted in the seminal application of phi-value analysis to the study of barnase, a bacterial RNAse used in many protein folding studies. Development of phi value analysis in combination with extensive protein engineering enabled an understanding of the kinetic intermediates during protein folding of barnase. In subsequent postdoctoral work at the University of Basel, he studied how mitochondria refold proteins after importing them. In 1996, he moved to Northwestern University. In 2012, he moved to The University of Texas at Austin.

Natalie C. J. Strynadka FRS is a professor of Biochemistry in the Department of Biochemistry and Molecular Biology at the University of British Columbia.