Related Research Articles
Lafora disease is a rare, adult-onset and autosomal recessive genetic disorder which results in myoclonus epilepsy and usually results in death several years after the onset of symptoms. The disease is characterized by the accumulation of inclusion bodies, known as Lafora bodies, within the cytoplasm of the cells in the heart, liver, muscle, and skin. Lafora disease is also a neurodegenerative disease that causes impairment in the development of brain (cerebral) cortical neurons and is a glycogen metabolism disorder.
A hypnic jerk, hypnagogic jerk, sleep start, sleep twitch, myoclonic jerk, or night start is a brief and sudden involuntary contraction of the muscles of the body which occurs when a person is beginning to fall asleep, often causing the person to jump and awaken suddenly for a moment. Hypnic jerks are one form of involuntary muscle twitches called myoclonus.
Absence seizures are one of several kinds of generalized seizures. In the past, absence epilepsy was referred to as "pyknolepsy," a term derived from the Greek word "pyknos," signifying "extremely frequent" or "grouped".These seizures are sometimes referred to as petit mal seizures ; however, usage of this terminology is no longer recommended. Absence seizures are characterized by a brief loss and return of consciousness, generally not followed by a period of lethargy. Absence seizures are most common in children. They affect both sides of the brain.

Myoclonus is a brief, involuntary, irregular twitching of a muscle, a joint, or a group of muscles, different from clonus, which is rhythmic or regular. Myoclonus describes a medical sign and, generally, is not a diagnosis of a disease. These myoclonic twitches, jerks, or seizures are usually caused by sudden muscle contractions or brief lapses of contraction. The most common circumstance under which they occur is while falling asleep. Myoclonic jerks occur in healthy people and are experienced occasionally by everyone. However, when they appear with more persistence and become more widespread they can be a sign of various neurological disorders. Hiccups are a kind of myoclonic jerk specifically affecting the diaphragm. When a spasm is caused by another person it is known as a provoked spasm. Shuddering attacks in babies fall in this category.
Opsoclonus myoclonus syndrome (OMS), also known as opsoclonus-myoclonus-ataxia (OMA), is a rare neurological disorder of unknown cause which appears to be the result of an autoimmune process involving the nervous system. It is an extremely rare condition, affecting as few as 1 in 10,000,000 people per year. It affects 2 to 3% of children with neuroblastoma and has been reported to occur with celiac disease and diseases of neurologic and autonomic dysfunction.
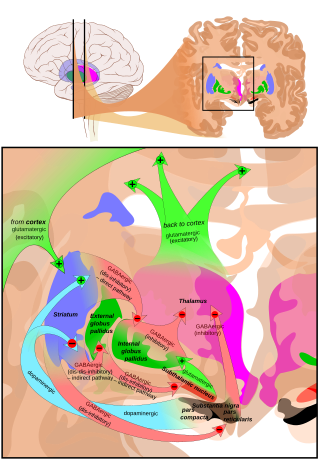
Hyperkinesia refers to an increase in muscular activity that can result in excessive abnormal movements, excessive normal movements, or a combination of both. Hyperkinesia is a state of excessive restlessness which is featured in a large variety of disorders that affect the ability to control motor movement, such as Huntington's disease. It is the opposite of hypokinesia, which refers to decreased bodily movement, as commonly manifested in Parkinson's disease.

Mucolipidosis type I is an inherited lysosomal storage disease that results from a deficiency of the enzyme alpha-N -acetyl neuraminidase (sialidase). The lack of this enzyme results in an abnormal accumulation of complex carbohydrates known as mucopolysaccharides, and of fatty substances known as mucolipids. Both of these substances accumulate in bodily tissues.
Myoclonic epilepsy refers to a family of epilepsies that present with myoclonus. When myoclonic jerks are occasionally associated with abnormal brain wave activity, it can be categorized as myoclonic seizure. If the abnormal brain wave activity is persistent and results from ongoing seizures, then a diagnosis of myoclonic epilepsy may be considered.

MERRF syndrome is a mitochondrial disease. It is extremely rare, and has varying degrees of expressivity owing to heteroplasmy. MERRF syndrome affects different parts of the body, particularly the muscles and nervous system. The signs and symptoms of this disorder appear at an early age, generally childhood or adolescence. The causes of MERRF syndrome are difficult to determine, but because it is a mitochondrial disorder, it can be caused by the mutation of nuclear DNA or mitochondrial DNA. The classification of this disease varies from patient to patient, since many individuals do not fall into one specific disease category. The primary features displayed on a person with MERRF include myoclonus, seizures, cerebellar ataxia, myopathy, and ragged red fibers (RRF) on muscle biopsy, leading to the disease's name. Secondary features include dementia, optic atrophy, bilateral deafness, peripheral neuropathy, spasticity, or multiple lipomata. Mitochondrial disorders, including MERRFS, may present at any age.
Juvenile myoclonic epilepsy (JME), also known as Janz syndrome or impulsive petit mal, is a form of hereditary, idiopathic generalized epilepsy, representing 5–10% of all epilepsy cases. Typically it first presents between the ages of 12 and 18 with myoclonic seizures. These events typically occur after awakening from sleep, during the evening or when sleep-deprived. JME is also characterized by generalized tonic–clonic seizures, and a minority of patients have absence seizures. It was first described by Théodore Herpin in 1857. Understanding of the genetics of JME has been rapidly evolving since the 1990s, and over 20 chromosomal loci and multiple genes have been identified. Given the genetic and clinical heterogeneity of JME some authors have suggested that it should be thought of as a spectrum disorder.

Ring chromosome 20, ring-shaped chromosome 20 or r(20) syndrome is a rare human chromosome abnormality where the two arms of chromosome 20 fuse to form a ring chromosome. The syndrome is associated with epileptic seizures, behaviour disorders and intellectual disability.
Unverricht–Lundborg disease is the most common form of an uncommon group of genetic epilepsy disorders called the progressive myoclonus epilepsies. It is caused due to a mutation in the cystatin B gene (CSTB). The disease is named after Heinrich Unverricht, who first described it in 1891, and Herman Bernhard Lundborg, who researched it in greater detail in 1901 and 1903. ULD onsets in children between the ages of 6 and 16; there are no known cases in which the person was older than 18. Most cases originate from the Baltic region of Europe, though many have been reported from countries in the Mediterranean.
Progressive Myoclonic Epilepsies (PME) are a rare group of inherited neurodegenerative diseases characterized by myoclonus, resistance to treatment, and neurological deterioration. The cause of PME depends largely on the type of PME. Most PMEs are caused by autosomal dominant or recessive and mitochondrial mutations. The location of the mutation also affects the inheritance and treatment of PME. Diagnosing PME is difficult due to their genetic heterogeneity and the lack of a genetic mutation identified in some patients. The prognosis depends largely on the worsening symptoms and failure to respond to treatment. There is no current cure for PME and treatment focuses on managing myoclonus and seizures through antiepileptic medication (AED).
Ramsay Hunt syndrome type 1 is a rare, degenerative, neurological disorder characterized by myoclonus epilepsy, intention tremor, progressive ataxia and occasionally cognitive impairment
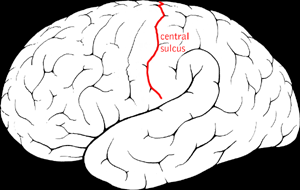
Benign Rolandic epilepsy or self-limited epilepsy with centrotemporal spikes is the most common epilepsy syndrome in childhood. Most children will outgrow the syndrome, hence the label benign. The seizures, sometimes referred to as sylvian seizures, start around the central sulcus of the brain.
Panayiotopoulos syndrome is a common idiopathic childhood-related seizure disorder that occurs exclusively in otherwise normal children and manifests mainly with autonomic epileptic seizures and autonomic status epilepticus. An expert consensus has defined Panayiotopoulos syndrome as "a benign age-related focal seizure disorder occurring in early and mid-childhood. It is characterized by seizures, often prolonged, with predominantly autonomic symptoms, and by an EEG [electroencephalogram] that shows shifting and/or multiple foci, often with occipital predominance."
Myoclonic astatic epilepsy (MAE), also known as myoclonic atonic epilepsy or Doose syndrome, is a generalized idiopathic epilepsy. It is characterized by the development of myoclonic seizures and/or myoclonic astatic seizures. Some of the common monogenic causes include mutations in the genes SLC6A1 (3p25.3),CHD2 (15q26.1), AP2M1 (10q23.2).
People with epilepsy may be classified into different syndromes based on specific clinical features. These features include the age at which seizures begin, the seizure types, and EEG findings, among others. Identifying an epilepsy syndrome is useful as it helps determine the underlying causes as well as deciding what anti-seizure medication should be tried. Epilepsy syndromes are more commonly diagnosed in infants and children. Some examples of epilepsy syndromes include benign rolandic epilepsy, childhood absence epilepsy and juvenile myoclonic epilepsy. Severe syndromes with diffuse brain dysfunction caused, at least partly, by some aspect of epilepsy, are also referred to as epileptic encephalopathies. These are associated with frequent seizures that are resistant to treatment and severe cognitive dysfunction, for instance Lennox-Gastaut syndrome and West syndrome.
A neonatal seizure is a seizure in a baby younger than age 4-weeks that is identifiable by an electrical recording of the brain. It is an occurrence of abnormal, paroxysmal, and persistent ictal rhythm with an amplitude of 2 microvolts in the electroencephalogram,. These may be manifested in form of stiffening or jerking of limbs or trunk. Sometimes random eye movements, cycling movements of legs, tonic eyeball movements, and lip-smacking movements may be observed. Alteration in heart rate, blood pressure, respiration, salivation, pupillary dilation, and other associated paroxysmal changes in the autonomic nervous system of infants may be caused due to these seizures. Often these changes are observed along with the observance of other clinical symptoms. A neonatal seizure may or may not be epileptic. Some of them may be provoked. Most neonatal seizures are due to secondary causes. With hypoxic ischemic encephalopathy being the most common cause in full term infants and intraventricular hemorrhage as the most common cause in preterm infants.

Solute carrier family 25 member 22 is a protein that in humans is encoded by the SLC25A22 gene. This gene encodes a mitochondrial glutamate carrier. Mutations in this gene are associated with early infantile epileptic encephalopathy. Expression of this gene is increased in colorectal tumor cells.
References
- 1 2 "Benign Neonatal Sleep Myoclonus: eMedicine Pediatrics: Cardiac Disease and Critical Care Medicine". 2 January 2015. Retrieved 31 May 2018.
- ↑ Di Capua, M; Fusco, L; Ricci, S; Vigevano, F (April 1993). "Benign neonatal sleep myoclonus: clinical features and video-polygraphic recordings". Mov Disord. 8 (2): 191–4. doi:10.1002/mds.870080213. PMID 8474488. S2CID 43231188 . Retrieved 13 May 2020.