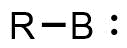Above: Upon reduction of the arylboron dichloride, a borylene is liberated. This intermediate adds into a C-C bond in the mesityl group. Below: Reduction of an dichloroaminoborane with Na/K yields a transient aminoborylene. Four equivalents of this species attack toluene to give a particularly complex product.
As discussed above, free borylenes have yet to be isolated, but they have been the subject of a number of computational studies and have investigated spectroscopically and experimentally. B-R (R=H, F, Cl, Br, I, NH2, C2H, Ph) have been observed via microwave or IR spectroscopy at low temperature via elaborate procedures.[13][14][15][16] When generated as reactive intermediates, borylenes have been shown to activate strong C-C single bonds, yielding products analogous to an organometallic oxidative addition reaction. Most commonly, these are generated via reduction of an organoborane dichloride, but photolysis of other boranes can also afford short-lived borylene species.
As might be expected, calculations have demonstrated that the HOMO is composed of the nonbonding electrons on boron (nσ-type, sp character). The LUMO and LUMO+1 are empty, orthogonal pπ-type orbitals and are degenerate in energy except in the case where R breaks the symmetry of the molecule, thus lifting the degeneracy. Unlike carbenes, which can exist in either singlet or triplet ground states, calculations have indicated that all yet-studied borylenes have a singlet ground spin state. The smallest singlet-triplet gap was calculated to be 8.2 kcal/mol for Me3Si-B. Aminoborylene (H2NB) is a slight exception to the above paradigm, as the nitrogen lone pair donates into an unoccupied boron p orbital. Thus, there is formally a double bond between boron and nitrogen; the π* combination of this interaction serves as the LUMO+1.[17]
Diborene B-B π-bonding HOMO.
Mono-Lewis base-stabilized borylenes
Above: Diborene dimer generated via reduction of an (NHC)borane adduct. Middle and bottom: two examples of mono-Lewis base-stabilized borylenes using CAAC and DAC ligands.
The first example of a borylene stabilized by a single Lewis base was reported in 2007 and exists as a dimer—a diborene. An (NHC)BBr3 adduct was reduced to generate a probable (NHC)B-H intermediate that subsequently dimerized to form the diborene. A similar species with a boron–boron single bond was also observed. The diborene has an incredibly short boron–boron bond length of 1.560(18) Å, further supporting the assignment of a double bond. DFT and NBO calculations were performed on a model system (with Dipp moieties replaced by H). Although some differences between the calculated and crystal structures were evident, they could primarily be ascribed to distortions from planarity caused by the bulky Dipp groups. The HOMO was calculated to be a B-B π-bonding orbital and the HOMO-1 is of mixed B-H and B-B σ-bonding character. NBO calculations supported the above assessments, as populations for the B-B σ- and π-bonding orbitals were calculated to be 1.943 and 1.382 respectively.[18]
Diborene B-B σ-bonding HOMO-1.
A number of similar compounds have been generated and isolated, and several studies involving putative mono-Lewis base-stabilized borylene intermediates have been reported. However, an isolable example remained elusive until 2014.[19] Betrand et al. argued that due to boron's electropositivity and thus preference to be electron-poor, CAAC (cyclic (alkyl)(amino)carbene) might serve as a better Lewis base than the more commonplace NHC.[21] The (NHC)borane adduct was prepared then reduced with Co(Cp*)2. One equivalent of reductant yielded an aminoboryl radical and a second reduction event lead to the desired (CAAC)borylene.[19] Another group followed a similar synthetic strategy using DAC(diamidocarbene); the reduction of a (DAC)borane derivative afforded an analogous (DAC)borylene (see figure).[20] Although the C=B=NR2 structure is similar in nature to aminoboraalkenes, an exploration of molecular orbitals gives an entirely different picture: as expected, the HOMO is a bond of π symmetry derived from the donation of boron's lone pair into the empty orbital on carbon. As previously discussed, a nitrogen lone pair donates into an empty boron p-orbital to form a π bond; the out of phase combination serves as a high-energy LUMO+2.[19]
Diborene B-B σ-bonding-derived NBO.Braunschweig's 2018 dinitrogen activation with a transient borylene speciesDiborene B lone pair-derived NBO.
The first example of dinitrogen fixation at a p-block element was published in 2018 by Holger Braunschweig et al., whereby one molecule of dinitrogen is bound by two transient mono-Lewis base-stabilized borylene species.[22] The resulting dianion was subsequently oxidized to a neutral compound, and reduced using water.
Bis-Lewis base-stabilized borylenes
DiLewisBaseBoryleneBis(CAAC)BH LUMO.
Taking inspiration from Robinson's above diborene synthesis,[21][18] Bertrand et al. swapped NHC for CAAC and successfully isolated the first bis-Lewis base-stabilized borylene in 2011.[24] Reduction of (CAAC)BBr3 with KC8 in the presence of excess CAAC afforded the bis(CAAC)BH. A labeling study indicated that the H-atom was abstracted from an aryl group associated with the CAAC. Reduction of (CAAC)BBr3 yields the same terminal borylene even in the absence of additional Lewis base via a mechanism that remains poorly understood.[24] Exploitation of this procedure has been used to form mixed bis-Lewis base-stabilized borylenes as well.[25] Several other routes have also been proposed. A more novel one employs methyl triflate to abstract a hydride from (CAAC)BH3. Treatment with a Lewis base, followed by triflic acid and KC8 afford the desired (CAAC)(Lewis base)BH.[26] Although the reported case uses only specific Lewis bases, the approach is argued to be highly generalizable.[21][26] A number of other compounds in this class have been generated using borylene-transition metal complexes as precursors. Treatment of (OC)5M=B-Tp with carbon monoxide or acetonitrile yields the corresponding adducts: (CO)2B-Tp and (MeNC)2B-Tp.[27]
Bis(CAAC)BH HOMO.
Bonding in these complexes is quite similar to that in mono-Lewis base compounds. At least one π-acceptor ligand is present in all known examples of these compounds, and the B-L bond strength tends to scale with the π-acidity of the Lewis base. Low-energy σ-donation orbitals from the base to boron are present in these compounds, and the π-interaction from boron's lone pair to the Lewis base serves as the HOMO. Calculated electronic structure for a number of borylene complexes were compared with their isoelectronic homologues: carbone complexes (CL2) and nitrogen cation complexes ((N+)L2).[28]
Borylene-transition metal complexes
The first transition metal complex reported by Braunschweig et al. featured a borylene ligand bridging between two manganese centers: [ μ-BX{η5-C5H4R}Mn(CO)2}2] (R=H, Me; X=NMe2).[29] The first terminal borylene complex [(CO)5MBN(SiMe3)2] was prepared by the same group several years later. Two previous structures – [(CO)4Fe(BNMe2)] and [(CO)4Fe{BN(SiMe3)2}] – had been proposed by other groups but disqualified due to inconsistent 11B-NMR data.[30] A number of diborylene complexes have also been described. The first of these, [(η5-C5Me5)Ir{BN(SiMe3)2}2], was prepared by the photochemical reaction of [(η5-C5Me5)Ir(CO)2] with [(OC)5Cr{BN(SiMe3)2}].[31] One unusual reaction exhibited by these complexes is coupling of borylene and carbon monoxide ligands. Catenation of an iron borylene complex has generate an iron complex of a tetraboron (B4) chain.[32]
Orbitally, the interactions between transition metals and borylenes tend to be similar to the above Lewis acids and borylenes. A number of computational studies have been performed on these systems. A sample paper from 2000 employed NBO to analyze a series of related complexes. Taking [(CO)4Fe{BN(SiH3)2}] as an example, it was calculated that—as expected—the boron moiety is relatively electron-poor (+0.59 charge). The Fe-B π-bonding orbitals were found to have populations of 0.39 and 0.48 whereas the σ-bonding had 0.61. Thus, the Wiberg bond index of the Fe-B bond was a relatively strong 0.65 (compare: Fe-CO was 0.62 in the same complex. The analogous tungsten complex had a bond index value of 0.82. Overall, the paper concludes that transition metal-borylene bonds are very strong. However, the bonding has strong ionic contributions. Orbital attractions are primarily σ- accompanied by weaker π-interactions. Unlike corresponding metal-carbyne complexes, the bond order in all studied cases was less than 1.[33]
References
↑ Braunschweig, H.; Colling, M. (2003). "The Chemistry of Borylene Complexes". Eur. J. Inorg. Chem. 2003 (3): 393–403. doi:10.1002/ejic.200390054.
↑ Braunschweig, H; Shang, R (2015). "Reactivity of transition-metal borylene complexes: recent advances in B-C and B-B bond formation via borylene ligand coupling". Inorg Chem. 54 (7): 3099–106. doi:10.1021/acs.inorgchem.5b00091. PMID25760461.
↑ Dahcheh, F.; Martin, D.; Stephan, D. W.; Bertrand, G. (2014). "Synthesis and Reactivity of a CAAC–Aminoborylene Adduct: A Hetero-Allene or an Organoboron Isoelectronic with Singlet Carbenes". Angewandte Chemie International Edition. 53 (48): 13159–13163. doi:10.1002/anie.201408371. PMID25267591.
↑ Braunschweig, Holger; Dewhurst, Rian D.; Hupp, Florian; Nutz, Marco; Radacki, Krzysztof; Tate, Christopher W.; Vargas, Alfredo (2015). "Multiple complexation of CO and related ligands to a main-group element". Nature. 522 (7556): 327–330. Bibcode:2015Natur.522..327B. doi:10.1038/nature14489. PMID26085273. S2CID4454142.
↑ Grigsby, Warren J.; Power, Philip P. (1996-01-01). "Isolation and Reduction of Sterically Encumbered Arylboron Dihalides: Novel Boranediyl Insertion into C−C σ-Bonds". Journal of the American Chemical Society. 118 (34): 7981–7988. doi:10.1021/ja960918j. ISSN0002-7863.
↑ Meller, Anton; Seebold, Uwe; Maringgele, Walter; Noltemeyer, Mathias; Sheldrick, George M. (1989-10-01). "Synthesis and structure of novel polycyclic species from toluene and m-xylene and the dehalogenation product of difluoro(diisopropylamino)borane". Journal of the American Chemical Society. 111 (21): 8299–8300. doi:10.1021/ja00203a052. ISSN0002-7863.
↑ Bettinger, Holger F. (2006-03-01). "Phenylborylene: Direct Spectroscopic Characterization in Inert Gas Matrices". Journal of the American Chemical Society. 128 (8): 2534–2535. doi:10.1021/ja0548642. ISSN0002-7863. PMID16492027.
↑ Andrews, Lester; Hassanzadeh, Parviz; Martin, Jan M. L.; Taylor, Peter R. (1993-06-01). "Pulsed laser evaporated boron atom reactions with acetylene. Infrared spectra and quantum chemical structure and frequency calculations for several novel organoborane BC2H2 and HBC2 molecules". The Journal of Physical Chemistry. 97 (22): 5839–5847. doi:10.1021/j100124a010. ISSN0022-3654.
↑ Timms, Peter L. (1973-04-01). "Chemistry of boron and silicon subhalides". Accounts of Chemical Research. 6 (4): 118–123. doi:10.1021/ar50064a002. ISSN0001-4842.
↑ Nomoto, Miho; Okabayashi, Toshiaki; Klaus, Thomas; Tanimoto, Mitsutoshi (1997). "Microwave spectroscopic study of the BBr molecule". Journal of Molecular Structure. 413–414: 471–476. Bibcode:1997JMoSt.413..471N. doi:10.1016/s0022-2860(97)00145-2.
1 2 3 4 5 6 7 Wang, Yuzhong; Quillian, Brandon; Wei, Pingrong; Wannere, Chaitanya S.; Xie, Yaoming; King, R. Bruce; Schaefer, Henry F.; Schleyer, Paul v. R.; Robinson, Gregory H. (2007-10-01). "A Stable Neutral Diborene Containing a BB Double Bond". Journal of the American Chemical Society. 129 (41): 12412–12413. doi:10.1021/ja075932i. ISSN0002-7863. PMID17887683.
1 2 3 4 Dahcheh, Fatme; Martin, David; Stephan, Douglas W.; Bertrand, Guy (2014-11-24). "Synthesis and Reactivity of a CAAC–Aminoborylene Adduct: A Hetero-Allene or an Organoboron Isoelectronic with Singlet Carbenes". Angewandte Chemie International Edition. 53 (48): 13159–13163. doi:10.1002/anie.201408371. ISSN1521-3773. PMID25267591.
1 2 Ledet, Anthony D.; Hudnall, Todd W. (2016-06-14). "Reduction of a diamidocarbene-supported borenium cation: isolation of a neutral boryl-substituted radical and a carbene-stabilized aminoborylene". Dalton Transactions. 45 (24): 9820–9826. doi:10.1039/c6dt00300a. ISSN1477-9234. PMID26843319.
↑ Arrowsmith, Merle; Auerhammer, Dominic; Bertermann, Rüdiger; Braunschweig, Holger; Bringmann, Gerhard; Celik, Mehmet Ali; Dewhurst, Rian D.; Finze, Maik; Grüne, Matthias (2016-11-07). "Generation of Dicoordinate Boron(I) Units by Fragmentation of a Tetra-Boron(I) Molecular Square". Angewandte Chemie International Edition. 55 (46): 14464–14468. doi:10.1002/anie.201608429. ISSN1521-3773. PMID27730749.
1 2 Ruiz, David A.; Melaimi, Mohand; Bertrand, Guy (2014-06-24). "An efficient synthetic route to stable bis(carbene)borylenes [(L1)(L2)BH]". Chem. Commun. 50 (58): 7837–7839. doi:10.1039/c4cc03497j. ISSN1364-548X. PMID24909943.
↑ Braunschweig, Holger; Dewhurst, Rian D.; Hupp, Florian; Nutz, Marco; Radacki, Krzysztof; Tate, Christopher W.; Vargas, Alfredo; Ye, Qing (June 2015). "Multiple complexation of CO and related ligands to a main-group element". Nature. 522 (7556): 327–330. Bibcode:2015Natur.522..327B. doi:10.1038/nature14489. ISSN1476-4687. PMID26085273. S2CID4454142.
↑ Celik, Mehmet Ali; Sure, Rebecca; Klein, Susanne; Kinjo, Rei; Bertrand, Guy; Frenking, Gernot (2012-04-27). "Borylene Complexes (BH)L2 and Nitrogen Cation Complexes (N+)L2: Isoelectronic Homologues of Carbones CL2". Chemistry – A European Journal. 18 (18): 5676–5692. doi:10.1002/chem.201103965. ISSN1521-3765. PMID22434609.
↑ Braunschweig, Holger; Wagner, Trixie (1995-04-13). "Synthesis and Structure of the First Transition Metal Borylene Complexes". Angewandte Chemie International Edition in English. 34 (7): 825–826. doi:10.1002/anie.199508251. ISSN1521-3773.
↑ Bertsch, Stefanie; Braunschweig, Holger; Christ, Bastian; Forster, Melanie; Schwab, Katrin; Radacki, Krzysztof (2010-12-03). "Towards Homoleptic Borylene Complexes: Incorporation of Two Borylene Ligands into a Mononuclear Iridium Species". Angewandte Chemie International Edition. 49 (49): 9517–9520. doi:10.1002/anie.201004103. ISSN1521-3773. PMID21053226.
↑ Braunschweig, Holger; Shang, Rong (2015-04-06). "Reactivity of Transition-Metal Borylene Complexes: Recent Advances in B–C and B–B Bond Formation via Borylene Ligand Coupling". Inorganic Chemistry. 54 (7): 3099–3106. doi:10.1021/acs.inorgchem.5b00091. ISSN0020-1669. PMID25760461.
↑ Uddin, Jamal; Boehme, Christian; Frenking, Gernot (2000-02-01). "Nature of the Chemical Bond between a Transition Metal and a Group-13 Element: Structure and Bonding of Transition Metal Complexes with Terminal Group-13 Diyl Ligands ER (E=B to Tl; R=Cp, N(SiH3)2, Ph, Me)". Organometallics. 19 (4): 571–582. doi:10.1021/om990936k. ISSN0276-7333.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.