Organoboron chemistry or organoborane chemistry studies organoboron compounds, also called organoboranes. These chemical compounds combine boron and carbon; typically, they are organic derivatives of borane (BH3), as in the trialkyl boranes.[1][2]
Organoboranes and -borates enable many chemical transformations in organic chemistry — most importantly, hydroboration and carboboration. Most reactions transfer a nucleophilic boron substituent to an electrophilic center either inter- or intramolecularly. In particular, α,β-unsaturated borates and borates with an α leaving group are highly susceptible to intramolecular 1,2-migration of a group from boron to the electrophilic α position. Oxidation or protonolysis of the resulting organoboranes generates many organic products, including alcohols, carbonyl compounds, alkenes, and halides.[3]
Properties of the B-C bond
The C-B bond has low polarity (electronegativity 2.55 for carbon and 2.04 for boron). Alkyl boron compounds are in general stable, though easily oxidized.
Structure of a rare monomeric boron hydride, R = i-Pr.
The most-studied class of organoboron compounds has the formula BRnH3−n. These compounds are catalysts, reagents, and synthetic intermediates. Except a few bulky derivatives, the primary and secondary hydrides (n = 1 or 2) are, like diborane itself, strongly Lewis acidic and dimerize in condensed phases. The trialkyl and triaryl derivatives, e.g. triethylboron, are typically only weakly Lewis acidic, and form monomers with a trigonal, planar boron center.[5]
Monoalkyl boranes are relatively rare. When the alkyl group is small, such as methyl, monoalkylboranes often redistribute to mixtures of diborane and di- and trialkylboranes. One example of an isolable (bulky) primary borane is thexylborane (ThxBH2), produced by the hydroboration of tetramethylethylene:[6] A chiral example is monoisopinocampheylborane, obtained by hydroboration of (−)‐α‐pinene with borane dimethyl sulfide. Although often written as IpcBH2, it is a dimer, [IpcBH2]2.[7]
Dialkylboranes are also rare with small alkyls. One common preparation reduces dialkylhalogenoboranes with metal hydrides.[8] An important application in organic synthesis is transmetallation to form organozinc compounds.[9][10] Nevertheless, some diaryl and dialkylboranes are well known. Dimesitylborane is a dimer (C6H2Me3)4B2H2) that reacts only slowly with simple terminal alkenes. It adds to alkynes to give alkenylboranes.[11] A hindered dialkylborane is disiamylborane, abbreviated Sia2BH, also a dimer. Owing to its steric bulk, it selectively hydroborates less hindered, usually terminal alkenes in the presence of more substituted alkenes.[12] Disiamylborane must be freshly prepared as its solutions can only be stored at 0°C for a few hours. Dicyclohexylborane Chx2BH exhibits improved thermal stability than Sia2BH.
A versatile dialkylborane is 9-BBN. Also called "banana borane", it exists as a dimer. It can be distilled without decomposition at 195°C (12mm Hg). Reactions with 9-BBN typically occur at 60–80°C, with most alkenes reacting within one hour. Tetrasubstituted alkenes add 9-BBN at elevated temperature. Hydroboration of alkenes with 9-BBN proceeds with excellent regioselectivity. It is more sensitive to steric differences than Sia2BH, perhaps because of it rigid C8 backbone. 9-BBN is more reactive towards alkenes than alkynes.[13]
Oxyacids and esters
Compounds of the type BRn(OR)3-n are called borinic esters (n = 2), boronic esters (n = 1), and borates (n = 0). Boronic acids are key to the Suzuki reaction. Trimethyl borate, debatably not an organoboron compound, is an intermediate in sodium borohydride production.
Adducts
Boranes and borinic, boronic, and borate esters all form adducts with appropriate Lewis bases.
Strong bases do not deprotonate boranes of the form R2BH. Instead these reactions afford the octet-complete adduct R2HB-base.[14]
Boron is renowned for cluster species, e.g. dodecaborate [B12H12]2-. Such clusters have many organic derivatives. One example is [B12(CH3)12]2- and its radical derivative [B12(CH3)12]−.[16] Related cluster compounds with carbon vertices are carboranes; the best known is orthocarborane, C2B10H12. Carboranes have few commercial applications. Anionic derivatives such as [C2B9H11]2−, called dicarbollides, ligate similarly to cyclopentadienide.
Borane cluster structures are built from the triangular (BR)3 unit, which is almost unknown in isolation. However, the corresponding aromatic dianion, (BR)2− 3, forms from careful dehalogenation of a RNBCl2 species.[17]
Organometallic compounds with metal-boron bonds (M–BR2) are boryl complexes, corresponding to the notional boryl anion R2B−, although the latter cannot be produced through deprotonation (see §Adducts). In one synthesis, the boryl anion moiety arose through lithium-halogen exchange:
Alkylideneboranes (RB=CRR) with a boron–carbon double bond are rare. One example, HB=CH2, can be detected at low temperature. The derivative CH3B=C(SiMe3)2 is fairly stable, but prone to cyclodimerisation.[20]
Some boron-substituted heterocycles are aromatic, but very few such arenes are stable. In borabenzene, boron replaces one CH center in benzene. Borabenzene and derivatives invariably appear as adducts, e.g., C5H5B-pyridine. The cyclic compound borole, a structural analog of pyrrole, has not been isolated, but substituted derivatives (boroles) are known. The cyclic compound borepin has been isolated and is aromatic.
Boron-boron multiple bonds are rare, although doubly-bonded dianions have been known since the 1990s.[21] Neutral analogues use NHC adducts, such as the following diborane(2) derivative:[22][23]
Each boron atom has an attached proton and is coordinated to a NHC carbene.[24][25]
Hydroboration with borane (BH3) equivalents converts only 33% of the starting olefin to product — boron-containing byproducts consume the remainder. The chelate effect improves that ratio for cyclic boron-containing reagents. One common cyclic organoboron reagent is 9-BBN.[30][31]
Metal-catalyzed borylation reactions produce an organoboron compound from aliphatic or aromatic C-H sigma bonds via a transition-metal catalyst. A common reagent is bis(pinacolato)diboron.
From other boron compounds
Carbon monoxide reacts with alkylboranes to form an unstable borane carbonyl. Then an alkyl substituent migrates from boron to the carbonyl carbon. For example, homologated primary alcohols result from organoboranes, carbon monoxide, and a reducing agent (here, sodium borohydride):[32]
Alkenylboranes
Alkynylboranes attack electrophiles to give trans alkenylboranes,[33] as in the first step of this olefin synthesis:
Reactions
Overall synthetic routes via organoboron compounds
The key property of organoboranes (R3B) and borates (R4B−, generated via addition of R− to R3B) is their susceptibility to reorganization. These compounds possess boron–carbon bonds polarized toward carbon. The boron-attached carbon is nucleophilic;[34] in borates, the nucleophicity suffices for intermolecular transfer to an electrophile.[35][3]
Boranes alone are generally not nucleophilic enough to transfer an R group intermolecularly. Instead, the group 1,2-migrates to an electrophilic carbon attached to boron, especially if that carbon is unsaturated or bears a good leaving group:[35]
An organic group's migration propensity depends on its ability to stabilize negative charge: alkynyl > aryl ≈ alkenyl > primary alkyl > secondary alkyl > tertiary alkyl.[36] Bis(norbornyl)borane and 9-BBN are often hydroboration reagents for this reason — only the hydroborated olefin is likely to migrate upon nucleophilic activation.
Migration retains configuration at the migrant carbon[37] and inverts it at the (presumably sp3-hybridized) terminus.[38] The resulting reorganized borane can then be oxidized or protolyzed to a final product.
Protonolysis
Organoboranes are unstable to Brønsted–Lowry acids, deboronating in favor of a proton. Consequently, organoboranes are easily removed from an alkane or alkene substrate, as in the second step of this olefin synthesis:[33]
Addition to halocarbonyls
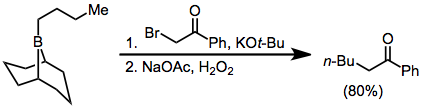
α-Halo enolates are common nucleophiles in borane reorganization. After nucleophilic attack at boron, the resulting ketoboronate eliminates the halogen and tautomerizes to a neutral enolborane. A functionalized carbonyl compound then results from protonolysis,[39] or quenching with other electrophiles:
Because the migration is stereospecific, this method synthesizes enantiopure α-alkyl or -aryl ketones.[40]
α-Haloester enolates add similarly to boranes, but with lower yields:[41]
Diazoesters and diazoketones remove the requirement for external base.[42] α,α'-Dihalo enolates react with boranes to form α-halo carbonyl compounds that can be further functionalized at the α position.[43]
Oxidation of an alkenylborane gives a boron-free enol.[47]
Halogenation
Organoborane activation with hydroxide or alkoxide and treatment with X2 yields haloalkanes. With excess base, two of the three alkyl groups attached to the boron atom may convert to halide, but disiamylborane permits only halogenation of the hydroborated olefin:[48]
Treatment of an alkenylborane with iodine or bromine induces migration of a boron-attached organic group. Alkynyl groups migrate selectively, forming enynes after treatment with sodium acetate and hydrogen peroxide:[49]
Homologated primary alcohols result from the treatment of organoboranes with carbon monoxide and a hydride:[51]
Tertiary alcohols with two identical groups attached to the alcohol carbon may be synthesized through an alkynylborane double migration:[47]
Carbonyl groups
Organoborates anions reductively eliminate against acyl halides. Here, the borate was generated from tri(cyclopentyl)borane and phenyllithium; the three cyclopentyl groups do not significantly migrate:[52]
Applications
Organoboron chemistry is mainly of commercial value in the pharmaceutical industry.
↑Bartlett, RuthA.; Dias, H.V.Rasika; Olmstead, MarilynM.; Power, PhilipP.; Weese, KennethJ. (1990). "Synthesis of the monomeric HBtrip2 (Trip - 2,4,6-iso-Pr3C6H2) and the x-ray crystal structures of [HBMes2]2 (Mes = 2,4,6,-Me3C6H2) and HBtrip2". Organometallics. 9: 146–150. doi:10.1021/om00115a023.
↑Brown, H.C. Organic Syntheses via Boranes John Wiley& Sons,Inc. New York: 1975. ISBN0-471-11280-1.
↑Negishi, Ei-Ichi; Brown, Herbert C. (1974). "Thexylborane-A Highly Versatile Reagent for Organic Synthesis via Hydroboration". Synthesis. 1974 (2): 77–89. doi:10.1055/s-1974-23248.
↑Dhar, Raj K.; Josyula, Kanth V. B.; Todd, Robert; Gagare, Pravin D.; Ramachandran, Veeraraghavan (2001). "Diisopinocampheylborane". Encyclopedia of Reagents for Organic Synthesis. pp.1–10. doi:10.1002/047084289X.rd248.pub3. ISBN9780470842898.
↑Brown, H. C.; Kulkarni, S. U. (1981). "Organoboranes: XXV. Hydridation of dialkylhaloboranes. New practical syntheses of dialkylboranes under mild conditions". Journal of Organometallic Chemistry. 218: 299. doi:10.1016/S0022-328X(00)81001-3.
↑Hupe, E.; Knochel, P. (2001). "Stereoselective Synthesis of Secondary Organozinc Reagents and Their Reaction with Heteroatomic Electrophiles". Organic Letters. 3 (1): 127–30. doi:10.1021/ol0068400. PMID11429854.
↑Pelter, A.; Singaram, S.; Brown, H. C. (1983). "The Dimesitylboron Group in Organic Chemistry. 6 Hydroborations with dimesitylborane". Tetrahedron Letters. 24 (13): 1433. doi:10.1016/S0040-4039(00)81675-5.
↑Dodd, D.S.; Ochlschlager, A. C. (1992). "Synthesis of inhibitors of 2,3-oxidosqualene-lanosterol cyclase: conjugate addition of organocuprates to N-(carbobenzyloxy)-3-carbomethoxy-5,6-dihydro-4-pyridone". Journal of Organic Chemistry. 57 (10): 2794. doi:10.1021/jo00036a008.
↑Dhillon, R. S. (2007). Hydroboration and Organic Synthesis: 9-Borabicyclo [3.3.1] Nonane (9-BBN). Springer.
↑Hall, DennisG. Boronic Acids: Preparation, Applications in Organic Synthesis and Medicine. ISBN3-527-30991-8
↑Curran, D.P.; Solovyev, A.; Makhlouf, BrahmiM.; Fensterbank, L.; Malacria, M.; Lacôte, E. (2011). "Synthesis and Reactions of N-Heterocyclic Carbene Boranes". Angewandte Chemie International Edition. 50 (44): 10294–10317. doi:10.1002/anie.201102717. PMID21898724.
↑Grimes, R.N. (2016). Carboranes (3rded.). New York: Academic Press. ISBN9780128019054.
↑Kupfer, Thomas; Braunschweig, Holger; Radacki, Krzysztof (2015-12-07). "The Triboracyclopropenyl Dianion: The Lightest Possible Main-Group-Element Hückel π Aromatic". Angewandte Chemie International Edition. 54 (50): 15084–15088. doi:10.1002/anie.201508670. PMID26530854.
↑Power, PhilipP. (2003) [19 July 2002]. "Persistent and stable radicals of the heavier main group elements and related species". Chemical Reviews. 103 (3). American Chemical Society: 792. doi:10.1021/cr020406p.
↑Nicolaou, K.C.; Sarabia, F.; Ninkovic, S.; Finlay, M.R.V.; Boddy, C.N.C. (1998). "Probing the Ring Size of Epothilones: Total Synthesis of 14-, 15-, 17-, and 18 Epothilones A". Angewandte Chemie International Edition in English. 37 (1–2): 81–84. doi:10.1002/(sici)1521-3773(19980202)37:1/2<81::aid-anie81>3.0.co;2-c.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.