Alkene/alkyne oligomerizations
Nickel compounds catalyze the oligomerization of alkenes and alkynes. This property validated the research and development of Ziegler–Natta catalysts in the 1950s. That discovery shown by nickel impurities originating from an autoclave which killed the propagation reaction (Aufbau) in favor of termination reaction to a terminal alkene: the polymerization of ethylene suddenly stopped at 1-butene. This so-called nickel effect prompted the search for other catalysts capable of this reaction, with results in the finding of new catalysts that technically produced high molar mass polymers, like the modern Ziegler–Natta catalysts.
One practical implementation of alkyne oligomerization is the Reppe synthesis; for example in the synthesis of cyclooctatetraene:
-
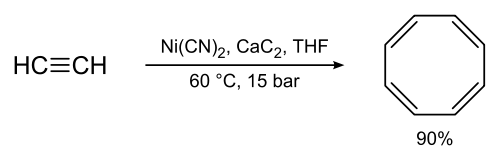
This is a formal [2+2+2+2]cycloaddition. The oligomerization of butadiene with ethylene to trans-1,4-hexadiene was an industrial process at one time.
Formal [2+2+2]cycloadditions also take place in alkyne trimerisation. This extensible trimerisation can generally include benzyne. [10] Benzyne is generated in situ from a benzene compound attached to a triflate and a trimethylsilyl substituent in the ortho- positions and reacts with a di-yne such as 1,7-octadiyne along with a nickel(II) bromide / zinc catalyst system (NiBr2 bis(diphenylphosphino) ethane / Zn) to synthesize the corresponding naphthalene derivative.

In the catalytic cycle elementary zinc serves to reduce nickel(II) to nickel(0) to which can then coordinate two alkyne bonds. A cyclometalation step follows to the nickelcyclopentadiene intermediate and then coordination of the benzyne which gives a C-H insertion reaction to the nickelcycloheptatriene compound. Reductive elimination liberates the tetrahydroanthracene compound.
The formation of organonickel compounds in this type of reaction is not always obvious but in a carefully designed experiment two such intermediates are formed quantitatively: [11] [12]
-

It is noted in one study [13] that this reaction only works with acetylene itself or with simple alkynes due to poor regioselectivity. From a terminal alkyne 7 isomers are possibly differing in the position of the substituents or the double bond positions. One strategy to remedy this problem employs certain diynes:
-

The selected reaction conditions also minimize the amount formed of competing [2+2+2]cycloaddition product to the corresponding substituted arene.

![Synthesis of [(TMEDA)Ni(o-tolyl)Cl]. Doyle reduction.tif](http://upload.wikimedia.org/wikipedia/commons/thumb/0/05/Doyle_reduction.tif/lossless-page1-350px-Doyle_reduction.tif.png)








