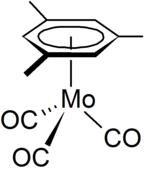Organometallic chemistry is the study of organometallic compounds, chemical compounds containing at least one chemical bond between a carbon atom of an organic molecule and a metal, including alkali, alkaline earth, and transition metals, and sometimes broadened to include metalloids like boron, silicon, and selenium, as well. Aside from bonds to organyl fragments or molecules, bonds to 'inorganic' carbon, like carbon monoxide, cyanide, or carbide, are generally considered to be organometallic as well. Some related compounds such as transition metal hydrides and metal phosphine complexes are often included in discussions of organometallic compounds, though strictly speaking, they are not necessarily organometallic. The related but distinct term "metalorganic compound" refers to metal-containing compounds lacking direct metal-carbon bonds but which contain organic ligands. Metal β-diketonates, alkoxides, dialkylamides, and metal phosphine complexes are representative members of this class. The field of organometallic chemistry combines aspects of traditional inorganic and organic chemistry.

In organic chemistry, olefin metathesis is an organic reaction that entails the redistribution of fragments of alkenes (olefins) by the scission and regeneration of carbon-carbon double bonds. Because of the relative simplicity of olefin metathesis, it often creates fewer undesired by-products and hazardous wastes than alternative organic reactions. For their elucidation of the reaction mechanism and their discovery of a variety of highly active catalysts, Yves Chauvin, Robert H. Grubbs, and Richard R. Schrock were collectively awarded the 2005 Nobel Prize in Chemistry.
A transition metal carbene complex is an organometallic compound featuring a divalent organic ligand. The divalent organic ligand coordinated to the metal center is called a carbene. Carbene complexes for almost all transition metals have been reported. Many methods for synthesizing them and reactions utilizing them have been reported. The term carbene ligand is a formalism since many are not derived from carbenes and almost none exhibit the reactivity characteristic of carbenes. Described often as M=CR2, they represent a class of organic ligands intermediate between alkyls (−CR3) and carbynes (≡CR). They feature in some catalytic reactions, especially alkene metathesis, and are of value in the preparation of some fine chemicals.

Alkyne metathesis is an organic reaction that entails the redistribution of alkyne chemical bonds. The reaction requires metal catalysts. Mechanistic studies show that the conversion proceeds via the intermediacy of metal alkylidyne complexes. The reaction is related to olefin metathesis.
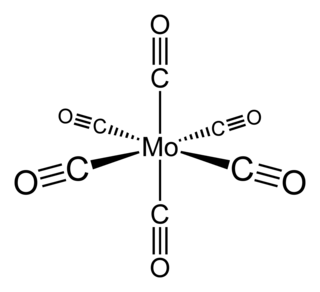
Molybdenum hexacarbonyl (also called molybdenum carbonyl) is the chemical compound with the formula Mo(CO)6. This colorless solid, like its chromium, tungsten, and seaborgium analogues, is noteworthy as a volatile, air-stable derivative of a metal in its zero oxidation state.

Richard Royce Schrock is an American chemist and Nobel laureate recognized for his contributions to the olefin metathesis reaction used in organic chemistry.
Ring-closing metathesis (RCM) is a widely used variation of olefin metathesis in organic chemistry for the synthesis of various unsaturated rings via the intramolecular metathesis of two terminal alkenes, which forms the cycloalkene as the E- or Z- isomers and volatile ethylene.

Titanocene dichloride is the organotitanium compound with the formula (η5-C5H5)2TiCl2, commonly abbreviated as Cp2TiCl2. This metallocene is a common reagent in organometallic and organic synthesis. It exists as a bright red solid that slowly hydrolyzes in air. It shows antitumour activity and was the first non-platinum complex to undergo clinical trials as a chemotherapy drug.

Dicobalt octacarbonyl is an organocobalt compound with composition Co2(CO)8. This metal carbonyl is used as a reagent and catalyst in organometallic chemistry and organic synthesis, and is central to much known organocobalt chemistry. It is the parent member of a family of hydroformylation catalysts. Each molecule consists of two cobalt atoms bound to eight carbon monoxide ligands, although multiple structural isomers are known. Some of the carbonyl ligands are labile.

Tungsten hexacarbonyl (also called tungsten carbonyl) is an organometallic compound with the formula W(CO)6. This complex gave rise to the first example of a dihydrogen complex.

In organometallic chemistry, a metallacycle is a derivative of a carbocyclic compound wherein a metal has replaced at least one carbon center; this is to some extent similar to heterocycles. Metallacycles appear frequently as reactive intermediates in catalysis, e.g. olefin metathesis and alkyne trimerization. In organic synthesis, directed ortho metalation is widely used for the functionalization of arene rings via C-H activation. One main effect that metallic atom substitution on a cyclic carbon compound is distorting the geometry due to the large size of typical metals.
Organochromium chemistry is a branch of organometallic chemistry that deals with organic compounds containing a chromium to carbon bond and their reactions. The field is of some relevance to organic synthesis. The relevant oxidation states for organochromium complexes encompass the entire range of possible oxidation states from –4 (d10) in Na4[Cr–IV(CO)4] to +6 (d0) in oxo-alkyl complexes like Cp*CrVI(=O)2Me.
Organoiron chemistry is the chemistry of iron compounds containing a carbon-to-iron chemical bond. Organoiron compounds are relevant in organic synthesis as reagents such as iron pentacarbonyl, diiron nonacarbonyl and disodium tetracarbonylferrate. While iron adopts oxidation states from Fe(−II) through to Fe(VII), Fe(IV) is the highest established oxidation state for organoiron species. Although iron is generally less active in many catalytic applications, it is less expensive and "greener" than other metals. Organoiron compounds feature a wide range of ligands that support the Fe-C bond; as with other organometals, these supporting ligands prominently include phosphines, carbon monoxide, and cyclopentadienyl, but hard ligands such as amines are employed as well.
Organorhenium chemistry describes the compounds with Re−C bonds. Because rhenium is a rare element, relatively few applications exist, but the area has been a rich source of concepts and a few useful catalysts.

(Mesitylene)molybdenum tricarbonyl is an organomolybdenum compound derived from the aromatic compound mesitylene (1,3,5-trimethylbenzene) and molybdenum carbonyl. It exists as pale yellow crystals, which are soluble in organic solvents but decompose when in solution. It has been examined as a catalyst and reagent.
Transition metal carbyne complexes are organometallic compounds with a triple bond between carbon and the transition metal. This triple bond consists of a σ-bond and two π-bonds. The HOMO of the carbyne ligand interacts with the LUMO of the metal to create the σ-bond. The two π-bonds are formed when the two HOMO orbitals of the metal back-donate to the LUMO of the carbyne. They are also called metal alkylidynes—the carbon is a carbyne ligand. Such compounds are useful in organic synthesis of alkynes and nitriles. They have been the focus on much fundamental research.
A metal carbido complex is a coordination complex that contains a carbon atom as a ligand. They are analogous to metal nitrido complexes. Carbido complexes are a molecular subclass of carbides, which are prevalent in organometallic and inorganic chemistry. Carbido complexes represent models for intermediates in Fischer–Tropsch synthesis, olefin metathesis, and related catalytic industrial processes. Ruthenium-based carbido complexes are by far the most synthesized and characterized to date. Although, complexes containing chromium, gold, iron, nickel, molybdenum, osmium, rhenium, and tungsten cores are also known. Mixed-metal carbides are also known.
Organoniobium chemistry is the chemistry of compounds containing niobium-carbon (Nb-C) bonds. Compared to the other group 5 transition metal organometallics, the chemistry of organoniobium compounds most closely resembles that of organotantalum compounds. Organoniobium compounds of oxidation states +5, +4, +3, +2, +1, 0, -1, and -3 have been prepared, with the +5 oxidation state being the most common.

In coordination chemistry and organometallic chemistry, transition metal imido complexes is a coordination compound containing an imido ligand. Imido ligands can be terminal or bridging ligands. The parent imido ligand has the formula NH, but most imido ligands have alkyl or aryl groups in place of H. The imido ligand is generally viewed as a dianion, akin to oxide.
Carbonyl olefin metathesis is a type of metathesis reaction that entails, formally, the redistribution of fragments of an alkene and a carbonyl by the scission and regeneration of carbon-carbon and carbon-oxygen double bonds respectively. It is a powerful method in organic synthesis using simple carbonyls and olefins and converting them into less accessible products with higher structural complexity.