
Organotantalum chemistry is the chemistry of chemical compounds containing a carbon-to-tantalum chemical bond. A wide variety of compound have been reported, initially with cyclopentadienyl and CO ligands. Oxidation states vary from Ta(V) to Ta(-I).
Organotantalum chemistry is the chemistry of chemical compounds containing a carbon-to-tantalum chemical bond. A wide variety of compound have been reported, initially with cyclopentadienyl and CO ligands. Oxidation states vary from Ta(V) to Ta(-I).
![Structure of [Li(OEt2)3] [TaMe6] . POZJAP.png](http://upload.wikimedia.org/wikipedia/commons/thumb/c/c6/POZJAP.png/220px-POZJAP.png)
Pentamethyltantalum was reported by Richard Schrock in 1974. [1]
Salts of [Ta(CH3)6]− are prepared by alkylation of TaF5 using methyl lithium: [2]
Tantalum alkylidene complexes arise by treating trialkyltantalum dichloride with alkyl lithium reagents. This reaction initially forms a thermally unstable tetraalkyl-monochloro-tantalum complex, which undergoes α-hydrogen elimination, followed by alkylation of the remaining chloride. [1]
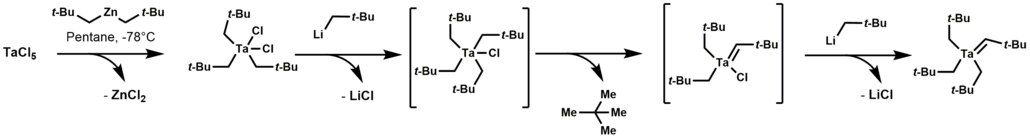
Tantalum alkylidene complexes are nucleophilic. [1] They effect a number of reactions including: olefinations, olefin metathesis, hydroaminoalkylation of olefins, and conjugate allylation of enones.

Ethylene, propylene, and styrene react with tantalum alkylidene complexes to yield olefin metathesis products. [3]
Some of the first reported organotantalum complexes were cyclopentadienyl derivatives. These arise from the salt metathesis reactions of sodium cyclopentadienide and tantalum pentachloride. An example of this is the first transition metal trihydride, Cp2TaH3. More soluble and better developed are derivatives of pentamethylcyclopentadiene such as Cp*TaCl4, Cp*2TaCl2, and Cp*2TaH3. [4]

Reduction of TaCl5 under an atmosphere of CO gives the salts of [Ta(CO)6]−. [5] These same anions can be obtained by carbonylation of tantalum arene complexes.
A number of tantalum isocyanide complexes are also known. [6]
Treatment of tantalum pentachloride with hexamethylbenzene (C6Me6), aluminium, and aluminium trichloride gives [M(η6-C6Me6)AlCl4]2. [7]
Tantalum-alkyne complexes [8] catalyze cyclotrimerizations. [9] [10] Some tantalum-alkyne complexes are precursors to allylic alcohols. [11] Tantalacyclopropenes are invoked as intermediates.

Organotantalum compounds are invoked as intermediates in C-alkylation of secondary amines with 1-alkenes using Ta(NMe2)5. [12] The chemistry developed by Maspero was later brought to fruition when Hartwig and Herzon reported the hydroaminoalkylation of olefins to form alkylamines: [13]

The catalytic cycle may proceed by β-hydrogen abstraction of the bisamide, which forms the metallaaziridine. Subsequent olefin insertion, protonolysis of the tantalum-carbon bond, and β-hydrogen abstraction affords the alkylamine product. [14] [15] [16]

Organotantalum reagents arise via transmetalation of organotin compounds with tantalum(V) chloride. [17] These organotantalum reagents promote the conjugate allylation of enones. Although the direct allylation of carbonyl groups is prevalent throughout the literature, little has been reported on the conjugate allylation of enones. [18]
Organotantalum compounds are of academic interest, but few or no commercial applications have been described.

In organic chemistry, an allyl group is a substituent with the structural formula −CH2−HC=CH2. It consists of a methylene bridge attached to a vinyl group. The name is derived from the scientific name for garlic, Allium sativum. In 1844, Theodor Wertheim isolated an allyl derivative from garlic oil and named it "Schwefelallyl". The term allyl applies to many compounds related to H2C=CH−CH2, some of which are of practical or of everyday importance, for example, allyl chloride.

In organic chemistry, olefin metathesis is an organic reaction that entails the redistribution of fragments of alkenes (olefins) by the scission and regeneration of carbon-carbon double bonds. Because of the relative simplicity of olefin metathesis, it often creates fewer undesired by-products and hazardous wastes than alternative organic reactions. For their elucidation of the reaction mechanism and their discovery of a variety of highly active catalysts, Yves Chauvin, Robert H. Grubbs, and Richard R. Schrock were collectively awarded the 2005 Nobel Prize in Chemistry.
A transition metal carbene complex is an organometallic compound featuring a divalent carbon ligand, itself also called a carbene. Carbene complexes have been synthesized from most transition metals and f-block metals, using many different synthetic routes such as nucleophilic addition and alpha-hydrogen abstraction. The term carbene ligand is a formalism since many are not directly derived from carbenes and most are much less reactive than lone carbenes. Described often as =CR2, carbene ligands are intermediate between alkyls (−CR3) and carbynes (≡CR). Many different carbene-based reagents such as Tebbe's reagent are used in synthesis. They also feature in catalytic reactions, especially alkene metathesis, and are of value in both industrial heterogeneous and in homogeneous catalysis for laboratory- and industrial-scale preparation of fine chemicals.
The Sakurai reaction is the chemical reaction of carbon electrophiles with allyltrimethylsilane catalyzed by strong Lewis acids. The reaction achieves results similar to the addition of an allyl Grignard reagent to the carbonyl.
A carbometallation is any reaction where a carbon-metal bond reacts with a carbon-carbon π-bond to produce a new carbon-carbon σ-bond and a carbon-metal σ-bond. The resulting carbon-metal bond can undergo further carbometallation reactions or it can be reacted with a variety of electrophiles including halogenating reagents, carbonyls, oxygen, and inorganic salts to produce different organometallic reagents. Carbometallations can be performed on alkynes and alkenes to form products with high geometric purity or enantioselectivity, respectively. Some metals prefer to give the anti-addition product with high selectivity and some yield the syn-addition product. The outcome of syn and anti- addition products is determined by the mechanism of the carbometallation.

Organocobalt chemistry is the chemistry of organometallic compounds containing a carbon to cobalt chemical bond. Organocobalt compounds are involved in several organic reactions and the important biomolecule vitamin B12 has a cobalt-carbon bond. Many organocobalt compounds exhibit useful catalytic properties, the preeminent example being dicobalt octacarbonyl.
Organoiron chemistry is the chemistry of iron compounds containing a carbon-to-iron chemical bond. Organoiron compounds are relevant in organic synthesis as reagents such as iron pentacarbonyl, diiron nonacarbonyl and disodium tetracarbonylferrate. Although iron is generally less active in many catalytic applications, it is less expensive and "greener" than other metals. Organoiron compounds feature a wide range of ligands that support the Fe-C bond; as with other organometals, these supporting ligands prominently include phosphines, carbon monoxide, and cyclopentadienyl, but hard ligands such as amines are employed as well.

Organomolybdenum chemistry is the chemistry of chemical compounds with Mo-C bonds. The heavier group 6 elements molybdenum and tungsten form organometallic compounds similar to those in organochromium chemistry but higher oxidation states tend to be more common.

Hydrogen auto-transfer, also known as borrowing hydrogen, is the activation of a chemical reaction by temporary transfer of two hydrogen atoms from the reactant to a catalyst and return of those hydrogen atoms back to a reaction intermediate to form the final product. Two major classes of borrowing hydrogen reactions exist: (a) those that result in hydroxyl substitution, and (b) those that result in carbonyl addition. In the former case, alcohol dehydrogenation generates a transient carbonyl compound that is subject to condensation followed by the return of hydrogen. In the latter case, alcohol dehydrogenation is followed by reductive generation of a nucleophile, which triggers carbonyl addition. As borrowing hydrogen processes avoid manipulations otherwise required for discrete alcohol oxidation and the use of stoichiometric organometallic reagents, they typically display high levels of atom-economy and, hence, are viewed as examples of Green chemistry.
In organic chemistry, carbonyl allylation describes methods for adding an allyl anion to an aldehyde or ketone to produce a homoallylic alcohol. The carbonyl allylation was first reported in 1876 by Alexander Zaitsev and employed an allylzinc reagent.

The Krische allylation involves the enantioselective iridium-catalyzed addition of an allyl group to an aldehyde or an alcohol, resulting in the formation of a secondary homoallylic alcohol. The mechanism of the Krische allylation involves primary alcohol dehydrogenation or, when using aldehyde reactants, hydrogen transfer from 2-propanol. Unlike other allylation methods, the Krische allylation avoids the use of preformed allyl metal reagents and enables the direct conversion of primary alcohols to secondary homoallylic alcohols.
Cobalt(II)–porphyrin catalysis is a process in which a Co(II) porphyrin complex acts as a catalyst, inducing and accelerating a chemical reaction.

Pentakis(dimethylamido)tantalum is an organometallic compound of tantalum. It is a colorless solid that is soluble in organic solvents. It hydrolyzes readily to release dimethylamine.
Organoniobium chemistry is the chemistry of compounds containing niobium-carbon (Nb-C) bonds. Compared to the other group 5 transition metal organometallics, the chemistry of organoniobium compounds most closely resembles that of organotantalum compounds. Organoniobium compounds of oxidation states +5, +4, +3, +2, +1, 0, -1, and -3 have been prepared, with the +5 oxidation state being the most common.
Pentamethyltantalum is a homoleptic organotantalum compound. It has a propensity to explode when it is melted. Its discovery was part of a sequence that led to Richard R. Schrock's Nobel Prize winning discovery in olefin metathesis.
Vinylcyclopropane [5+2] cycloaddition is a type of cycloaddition between a vinylcyclopropane (VCP) and an olefin or alkyne to form a seven-membered ring.
Jiro Tsuji was a Japanese chemist, notable for his discovery of organometallic reactions, including the Tsuji-Trost reaction, the Tsuji-Wilkinson decarbonylation, and the Tsuji-Wacker reaction.
Copper-catalyzed allylic substitutions are chemical reactions with unique regioselectivity compared to other transition-metal-catalyzed allylic substitutions such as the Tsuji-Trost reaction. They involve copper catalysts and "hard" carbon nucleophiles. The mechanism of copper-catalyzed allylic substitutions involves the coordination of copper to the olefin, oxidative addition and reductive elimination. Enantioselective versions of these reactions have been used in the synthesis of complex molecules, such as (R)-(-)-sporochnol and (S)-(-)-zearalenone.
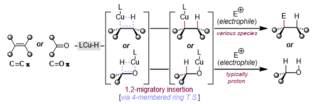
A hydrocupration is a chemical reaction whereby a ligated copper hydride species, reacts with a carbon-carbon or carbon-oxygen pi-system; this insertion is typically thought to occur via a four-membered ring transition state, producing a new copper-carbon or copper-oxygen sigma-bond and a stable (generally) carbon-hydrogen sigma-bond. In the latter instance (copper-oxygen), protonation (protodemetalation) is typical – the former (copper-carbon) has broad utility. The generated copper-carbon bond (organocuprate) has been employed in various nucleophilic additions to polar conjugated and non-conjugated systems and has also been used to forge new carbon-heteroatom bonds.
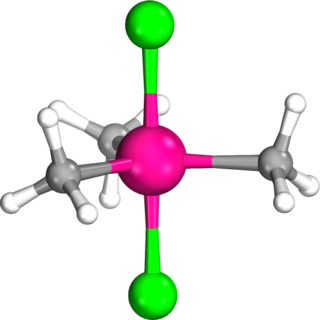
Dichlorotrimethyltantalum, or trimethyltantalum dichloride, is an organotantalum complex with the molecular formula TaCl2(CH3)3. It forms pale yellow, highly air- and water-sensitive crystals which easily sublime in vacuum at room temperature. It was the first reported σ-bonded alkyl complex of tantalum, synthesised by Gordon L. Juvinall at the California Institute of Technology in 1964. It serves as an important precursor for the preparation of a large number of Ta(V) complexes.
{{cite book}}: |journal= ignored (help)Compounds of carbon with other elements in the periodic table | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Legend |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||