Organocalcium chemistry is the chemistry of compounds containing a calcium to carbonbond,[1] or in broader definitions, organic compounds that contain calcium.[2] Although discovered around the same time as the now commonly utilized organomagnesium compounds,[3] organocalcium compounds were subject to greatly reduced interest due to drastic differences in stability. Because calcium metal is less reactive to organic reagents than magnesium[4] and the organocalcium compounds are more reactive than organomagnesium compounds, synthesis of novel compounds still poses a significant challenge. Calcium also has access to empty d orbitals that the lighter alkaline earth metals cannot access, and the degree to which this affects bonding and reactivity has sparked a fundamental debate.[5][6] Lastly, despite the inherent instability of most organocalcium complexes, the unique basicity and size of the calcium ion together with the highly polarized bonds formed has opened up applications for organocalcium compounds in organic transformations and catalytic cycles.
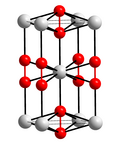
Structure of calcium carbide. Color code: red = C, gray = Ca.
In terms of scale (millions of kilograms) and practicality, calcium carbide is the dominant organocalcium compound. It is produced by reaction of carbon and calcium oxide:
CaO + 3 C → CaC2 + CO
It is used to prepare acetylene, which is widely used in welding. It reacts with nitrogen gas to give calcium cyanamide, a versatile synthetic intermediate.[7]
Compounds
In general, organocalcium synthesis is complicated by relatively unreactive calcium metal (compared to magnesium or the alkali metals due to a high atomization energy)[8][9] and high reactivity of most organocalcium compounds to oxygen, water, and even ethereal solvents.[10] To sustain the highly electropositive calcium center, the vast majority of compounds have anionic ligands by which they can be categorized, with neutral coordinating ligands utilized for increased stability.
History
Synthesis of phenylcalcium iodide-THF adduct and subsequent quenching to form biphenyl
In 1905 by Ernst Beckmann claimed the synthesis of phenylcalcium iodide by stirring of calcium shavings with iodobenzene in diethyl ether (Et2O).[3] Subsequent study by Henry Gilman and Ferdinand Schulze argued that the isolated product in this report was actually the Et2O adduct of CaI2,[11] and, although phenylcalcium halides have been reported numerous times,[12][13][14] they were usually characterized through subsequent derivatization products. It took a full century until, in 2005, Matthias Westerhausen and colleagues obtained the first structural characterization of an arylcalcium compound, crystallizing phenylcalcium iodide as an adduct of tetrahydrofuran (THF) and calcium oxide.[15] A consistent challenge in the formation of organocalcium compounds has been the activation of calcium metal. Mechanochemistry (ball-milling) has allowed the use of unactivated calcium.[16] The history of alkyl calcium compounds is also checkered until Lappert et al.'s synthesis of Ca[CHSi(CH3)3)]2]2.[17]
Lappert's synthesis of a dialkylcalcium compound.
Aryl, allyl, and alkyl derivatives
Structure of Ca3Me5I(ether)5.
Bis(allyl)calcium complexes are stabilized by sterically large, silyl substituents.[19] These syntheses use salt metathesis reactions, involving allyl potassium and CaI2. This strategy has been used to synthesize the unsubstituted complex Ca(η3-C3H5)2 as a soluble triglyme adduct.[20] and related species.[21] The carbon atom in the calcium-carbon bond takes on a significant negative charge. Because of the greater nucleophilicity of alkyl ligands, the alkylcalcium reagents are in general harder to synthesize than the arylcalcium compounds.[9] substituents to stabilize this negative charge. When targeting a Grignard analogue, the decreased reactivity from this method and the poor stability of the less protected methyl- and ethylcalcium halides has led to in situ generation of reactive alkylcalcium halides as the preferred method over the synthesis of isolable compounds.[22] Because of this poor stability, the pure organometallic dimethylcalcium was only isolated in 2018 by Reiner Anwander and colleagues as an insoluble, amorphous solid, with the THF adduct being structurally characterizable as a heptametallic cluster.[23]
Metallocenes
Demonstration of bent calcocene angle utilized for olefin binding with rough geometry taken from a crystal structure
The first synthesis of Cp2Ca (Cp = cyclopentadienyl) involved combining calcium metal and cyclopentadiene in THF, producing an insoluble, polymeric product.[24] According to X-ray crystallography calcocene is polymer.[25] This bent structure is observed in related compounds. For example, two butenyl-substituted Cp ligand will coordinate to Ca through both the five-membered rings and the olefins, in contrast to related magnesium compounds[26]
Low-oxidation-state compounds
Few organocalcium(I) compounds exist.[27][28] The first and only report of an isolable Ca(I) compound came in 2009, where two THF-coordinated Ca(I) ions sit on either side of an arene ring.[29] The π-antibonding orbitals of the sandwiched arene help stabilize the two calcium ions, which are further stabilized by the coordinating solvent. Other studies of Ca(I) were done at low temperatures in exotic conditions[30] or examine formally Ca(II) compounds that imply Ca(I)-containing intermediates either during synthesis or further reactivity.[31][32] A landmark example of this from Sjoerd Harder and coworkers is the reported reduction of arenes and N2 by a bridged Ca(I)-Ca(I) species generated in situ.[31] The ease of activating the normally inert N2 to turn it into a strong reductant even at room temperature highlights the instability of Ca(I) species. Although not isolable as a Ca(I)-Ca(I) dimer, it possesses similar reactivity as a stronger reducing agent than a Mg(I) dimer.
Synthesis of an inverse sandwich Ca(II) complex through an in situ generated Ca(I)-Ca(I) dimer (dipp = 2,6-diisopropylphenyl)
Bonding descriptions
The changes in properties going down the alkaline earth group causes calcium to possess qualitatively entirely distinct bonding characteristics than the lighter beryllium and magnesium ions. In particular, calcium is significantly larger, more reducing, and has a much lower electronegativity. This enforces a strong preference for the Ca(II) oxidation state and an essentially ionic bond with carbon, which can be reasonably described as a carbanion in the Ca-C bond.[27]
A key difference in calcium bonding descriptions compared to magnesium and beryllium is the occasional use of the unfilled 3d orbitals to fully explain bonding and structural patterns. For example, the bent nature of calcocene, and the potentially bent geometry of CaH2, can be explained by increased involvement of the 3d orbitals in bonding.[33][34] This has been highly debated, however, with other explanations invoking the polarizability of the larger Ca core[35] and a stabilizing van der Waals interaction between the two ligands.[36] A similar debate[5][6][37] is ongoing regarding the degree of π-backbonding in a Ca(CO)8 complex.[30] Although still controversial, computational studies on the degree of sp-d hybridization[38] have caused some to label Ca as an honorary transition metal.[37]
Reactivity
Heavy Grignard reactivity
Organocalcium compounds show some more similarities to organolithium chemistry over organomagnesium compounds. This is largely due to differences in electronegativity, which allow organocalcium compounds to function as a base more often than typical magnesium-based Grignard reagents do.[9] This basicity is exemplified by the facile deprotonation and subsequent cleavage of ethers such as THF.[39]
Organolithium compound-like deprotonation of THF and subsequent ether cleavage by an alkyl- or arylcalcium iodide
Another point of differentiation from the magnesium-based Grignard reagents is the higher positive charge localized on the calcium atom, due to the higher degree of ionicity in the Ca-C bond versus the Mg-C bond, which can enable unique reactivity not seen in the lighter alkaline earth compounds. For example, a dimeric Ca alkynide complex was shown to enable the coupling of two anionic alkynides to form an extended, fully double bonded four-carbon chain.[40] The previously mentioned in situ generation of reactive alkylcalcium species has also been successfully used to react with amines to form calcium amides.[22] This reactivity relies on fast ligand exchange of calcium Grignard reagents due to the ionic nature of this bond – the initially formed product is a heteroleptic calcium monoamide monohalide, but ligand exchange quickly forms the full calcium diamide and an insoluble calcium dihalide that drives the Schlenk equilibrium to completion. Non-Grignard alkylcalcium complexes have also shown unique reactivity, such as alkylation of benzene driven by the formation of a calcium hydride.[41]
In situ generated, unprotected alkylcalcium-based Grignard reagent used to generate a diamide organocalcium complex
Catalytic reactivity
Catalysis with organocalcium compounds has historically been limited and has never achieved any practicality. Multiple areas of catalysis have been investigated. Inspired by alkali metal-based organometallic compounds use in anionic polymerization, organocalcium compounds have also been investigated as polymerization catalysts.[2] For example, fast polymerization has been seen for polylactide synthesis with excellent selectivity for the isotactic form.[42] This is not only enabled by the previously discussed electronic and electrostatic differences, but also by the larger size of calcium in comparison to the alkali metals or magnesium. The larger size of calcium allows an unusual trigonal prismatic coordination geometry utilized throughout the mechanism.[43] The ionic nature of Ca-C bonding can also be leveraged for living polymerization, as was demonstrated for a stereoselective synthesis of polystyrene.[44]
First demonstration of intramolecular hydroamination catalyzed by a calcium amide by Michael Hill
Catalysis has also been performed using organocalcium compounds for a series of organic transformations. This most prominently includes hydroamination, where numerous viable substrates and modes of selectivity have been demonstrated.[2][45][46] Catalytic activity has also been shown for the analogous hydrophosphination,[47] the hydrogenation of alkene with dihydrogen,[48] regioselective hydrosilylation of conjugated alkenes,[49] and the hydroboration of alkenes, although the role of calcium in the latter mechanism is still debated.[50] The redistribution of arylsilane and hydrosilane groups has also been performed catalytically, relying on the cleavage and reformation of C-Si and Si-H bonds driven by the simultaneous cleavage and reformation of Ca-C and Ca-H bonds.[51][52]
Jenter, Jelena; Köppe, Ralf; Roesky, Peter W. (2011). "2,5-Bis{N-(2,6-diisopropylphenyl)iminomethyl}pyrrolyl Complexes of the Heavy Alkaline Earth Metals: Synthesis, Structures, and Hydroamination Catalysis". Organometallics. 30 (6): 1404–13. doi:10.1021/om100937c.
Arrowsmith, Merle; Crimmin, Mark R.; Barrett, Anthony G. M.; Hill, Michael S.; Kociok-Köhn, Gabriele; Procopiou, Panayiotis A. (2011). "Cation Charge Density and Precatalyst Selection in Group 2-Catalyzed Aminoalkene Hydroamination". Organometallics. 30 (6): 1493–1506. doi:10.1021/om101063m.
References
↑Massey, A. G. (2000). Main group chemistry. Inorganic chemistry (2nded.). Chichester; New York: Wiley. ISBN978-0-471-49037-1.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.