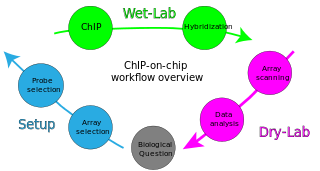
ChIP-on-chip is a technology that combines chromatin immunoprecipitation ('ChIP') with DNA microarray ("chip"). Like regular ChIP, ChIP-on-chip is used to investigate interactions between proteins and DNA in vivo. Specifically, it allows the identification of the cistrome, the sum of binding sites, for DNA-binding proteins on a genome-wide basis. Whole-genome analysis can be performed to determine the locations of binding sites for almost any protein of interest. As the name of the technique suggests, such proteins are generally those operating in the context of chromatin. The most prominent representatives of this class are transcription factors, replication-related proteins, like origin recognition complex protein (ORC), histones, their variants, and histone modifications.

Chromosome conformation capture techniques are a set of molecular biology methods used to analyze the spatial organization of chromatin in a cell. These methods quantify the number of interactions between genomic loci that are nearby in 3-D space, but may be separated by many nucleotides in the linear genome. Such interactions may result from biological functions, such as promoter-enhancer interactions, or from random polymer looping, where undirected physical motion of chromatin causes loci to collide. Interaction frequencies may be analyzed directly, or they may be converted to distances and used to reconstruct 3-D structures.

SOLiD (Sequencing by Oligonucleotide Ligation and Detection) is a next-generation DNA sequencing technology developed by Life Technologies and has been commercially available since 2006. This next generation technology generates 108 - 109 small sequence reads at one time. It uses 2 base encoding to decode the raw data generated by the sequencing platform into sequence data.
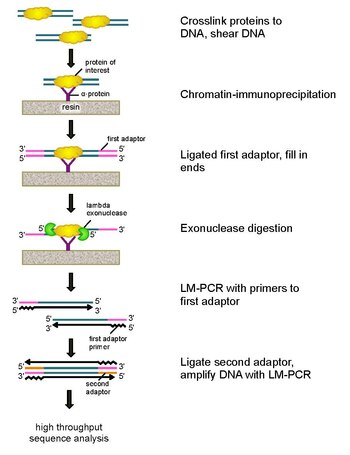
ChIP-sequencing, also known as ChIP-seq, is a method used to analyze protein interactions with DNA. ChIP-seq combines chromatin immunoprecipitation (ChIP) with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. It can be used to map global binding sites precisely for any protein of interest. Previously, ChIP-on-chip was the most common technique utilized to study these protein–DNA relations.
DNA adenine methyltransferase identification, often abbreviated DamID, is a molecular biology protocol used to map the binding sites of DNA- and chromatin-binding proteins in eukaryotes. DamID identifies binding sites by expressing the proposed DNA-binding protein as a fusion protein with DNA methyltransferase. Binding of the protein of interest to DNA localizes the methyltransferase in the region of the binding site. Adenine methylation does not occur naturally in eukaryotes and therefore adenine methylation in any region can be concluded to have been caused by the fusion protein, implying the region is located near a binding site. DamID is an alternate method to ChIP-on-chip or ChIP-seq.

RIP-chip is a molecular biology technique which combines RNA immunoprecipitation with a microarray. The purpose of this technique is to identify which RNA sequences interact with a particular RNA binding protein of interest in vivo. It can also be used to determine relative levels of gene expression, to identify subsets of RNAs which may be co-regulated, or to identify RNAs that may have related functions. This technique provides insight into the post-transcriptional gene regulation which occurs between RNA and RNA binding proteins.
Epigenomics is the study of the complete set of epigenetic modifications on the genetic material of a cell, known as the epigenome. The field is analogous to genomics and proteomics, which are the study of the genome and proteome of a cell. Epigenetic modifications are reversible modifications on a cell's DNA or histones that affect gene expression without altering the DNA sequence. Epigenomic maintenance is a continuous process and plays an important role in stability of eukaryotic genomes by taking part in crucial biological mechanisms like DNA repair. Plant flavones are said to be inhibiting epigenomic marks that cause cancers. Two of the most characterized epigenetic modifications are DNA methylation and histone modification. Epigenetic modifications play an important role in gene expression and regulation, and are involved in numerous cellular processes such as in differentiation/development and tumorigenesis. The study of epigenetics on a global level has been made possible only recently through the adaptation of genomic high-throughput assays.
Chromatin Interaction Analysis by Paired-End Tag Sequencing is a technique that incorporates chromatin immunoprecipitation (ChIP)-based enrichment, chromatin proximity ligation, Paired-End Tags, and High-throughput sequencing to determine de novo long-range chromatin interactions genome-wide.

Chromatin immunoprecipitation (ChIP) is a type of immunoprecipitation experimental technique used to investigate the interaction between proteins and DNA in the cell. It aims to determine whether specific proteins are associated with specific genomic regions, such as transcription factors on promoters or other DNA binding sites, and possibly define cistromes. ChIP also aims to determine the specific location in the genome that various histone modifications are associated with, indicating the target of the histone modifiers. ChIP is crucial for the advancements in the field of epigenomics and learning more about epigenetic phenomena.
H3K4me3 is an epigenetic modification to the DNA packaging protein Histone H3 that indicates tri-methylation at the 4th lysine residue of the histone H3 protein and is often involved in the regulation of gene expression. The name denotes the addition of three methyl groups (trimethylation) to the lysine 4 on the histone H3 protein.
Selective microfluidics-based ligand enrichment followed by sequencing (SMiLE-seq) is a technique developed for the rapid identification of DNA binding specificities and affinities of full length monomeric and dimeric transcription factors in a fast and semi-high-throughput fashion.
CUT&RUN sequencing, also known as cleavage under targets and release using nuclease, is a method used to analyze protein interactions with DNA. CUT&RUN sequencing combines antibody-targeted controlled cleavage by micrococcal nuclease with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. It can be used to map global DNA binding sites precisely for any protein of interest. Currently, ChIP-Seq is the most common technique utilized to study protein–DNA relations, however, it suffers from a number of practical and economical limitations that CUT&RUN sequencing does not.
H3K9me3 is an epigenetic modification to the DNA packaging protein Histone H3. It is a mark that indicates the tri-methylation at the 9th lysine residue of the histone H3 protein and is often associated with heterochromatin.
H3K79me2 is an epigenetic modification to the DNA packaging protein Histone H3. It is a mark that indicates the di-methylation at the 79th lysine residue of the histone H3 protein. H3K79me2 is detected in the transcribed regions of active genes.
CUT&Tag-sequencing, also known as cleavage under targets and tagmentation, is a method used to analyze protein interactions with DNA. CUT&Tag-sequencing combines antibody-targeted controlled cleavage by a protein A-Tn5 fusion with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. It can be used to map global DNA binding sites precisely for any protein of interest. Currently, ChIP-Seq is the most common technique utilized to study protein–DNA relations, however, it suffers from a number of practical and economical limitations that CUT&RUN and CUT&Tag sequencing do not. CUT&Tag sequencing is an improvement over CUT&RUN because it does not require cells to be lysed or chromatin to be fractionated. CUT&RUN is not suitable for single-cell platforms so CUT&Tag is advantageous for these.
ChIL sequencing (ChIL-seq), also known as Chromatin Integration Labeling sequencing, is a method used to analyze protein interactions with DNA. ChIL-sequencing combines antibody-targeted controlled cleavage by Tn5 transposase with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. It can be used to map global DNA binding sites precisely for any protein of interest. Currently, ChIP-Seq is the most common technique utilized to study protein–DNA relations, however, it suffers from a number of practical and economical limitations that ChIL-Sequencing does not. ChIL-Seq is a precise technique that reduces sample loss could be applied to single-cells.
H3K36me2 is an epigenetic modification to the DNA packaging protein Histone H3. It is a mark that indicates the di-methylation at the 36th lysine residue of the histone H3 protein.

MNase-seq, short for micrococcal nuclease digestion with deep sequencing, is a molecular biological technique that was first pioneered in 2006 to measure nucleosome occupancy in the C. elegans genome, and was subsequently applied to the human genome in 2008. Though, the term ‘MNase-seq’ had not been coined until a year later, in 2009. Briefly, this technique relies on the use of the non-specific endo-exonuclease micrococcal nuclease, an enzyme derived from the bacteria Staphylococcus aureus, to bind and cleave protein-unbound regions of DNA on chromatin. DNA bound to histones or other chromatin-bound proteins may remain undigested. The uncut DNA is then purified from the proteins and sequenced through one or more of the various Next-Generation sequencing methods.
H3K36me is an epigenetic modification to the DNA packaging protein Histone H3, specifically, the mono-methylation at the 36th lysine residue of the histone H3 protein.

Proximity ligation-assisted chromatin immunoprecipitation sequencing (PLAC-seq) is a chromatin conformation capture(3C)-based technique to detect and quantify genomic chromatin structure from a protein-centric approach. PLAC-seq combines in situ Hi-C and chromatin immunoprecipitation (ChIP), which allows for the identification of long-range chromatin interactions at a high resolution with low sequencing costs. Mapping long-range 3-dimensional(3D) chromatin interactions is important in identifying transcription enhancers and non-coding variants that can be linked to human diseases.