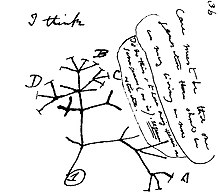
Cladistics is an approach to biological classification in which organisms are categorized in groups ("clades") based on hypotheses of most recent common ancestry. The evidence for hypothesized relationships is typically shared derived characteristics (synapomorphies) that are not present in more distant groups and ancestors. However, from an empirical perspective, common ancestors are inferences based on a cladistic hypothesis of relationships of taxa whose character states can be observed. Theoretically, a last common ancestor and all its descendants constitute a (minimal) clade. Importantly, all descendants stay in their overarching ancestral clade. For example, if the terms worms or fishes were used within a strict cladistic framework, these terms would include humans. Many of these terms are normally used paraphyletically, outside of cladistics, e.g. as a 'grade', which are fruitless to precisely delineate, especially when including extinct species. Radiation results in the generation of new subclades by bifurcation, but in practice sexual hybridization may blur very closely related groupings.
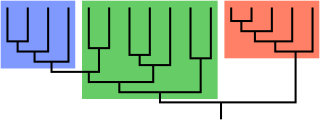
In biological phylogenetics, a clade, also known as a monophyletic group or natural group, is a grouping of organisms that are monophyletic – that is, composed of a common ancestor and all its lineal descendants – on a phylogenetic tree. In the taxonomical literature, sometimes the Latin form cladus is used rather than the English form. Clades are the fundamental unit of cladistics, a modern approach to taxonomy adopted by most biological fields.
In biology, phylogenetics is the study of the evolutionary history and relationships among or within groups of organisms. These relationships are determined by phylogenetic inference methods that focus on observed heritable traits, such as DNA sequences, protein amino acid sequences, or morphology. The result of such an analysis is a phylogenetic tree—a diagram containing a hypothesis of relationships that reflects the evolutionary history of a group of organisms.

A cladogram is a diagram used in cladistics to show relations among organisms. A cladogram is not, however, an evolutionary tree because it does not show how ancestors are related to descendants, nor does it show how much they have changed, so many differing evolutionary trees can be consistent with the same cladogram. A cladogram uses lines that branch off in different directions ending at a clade, a group of organisms with a last common ancestor. There are many shapes of cladograms but they all have lines that branch off from other lines. The lines can be traced back to where they branch off. These branching off points represent a hypothetical ancestor which can be inferred to exhibit the traits shared among the terminal taxa above it. This hypothetical ancestor might then provide clues about the order of evolution of various features, adaptation, and other evolutionary narratives about ancestors. Although traditionally such cladograms were generated largely on the basis of morphological characters, DNA and RNA sequencing data and computational phylogenetics are now very commonly used in the generation of cladograms, either on their own or in combination with morphology.

A phylogenetic tree, phylogeny or evolutionary tree is a graphical representation which shows the evolutionary history between a set of species or taxa during a specific time. In other words, it is a branching diagram or a tree showing the evolutionary relationships among various biological species or other entities based upon similarities and differences in their physical or genetic characteristics. In evolutionary biology, all life on Earth is theoretically part of a single phylogenetic tree, indicating common ancestry. Phylogenetics is the study of phylogenetic trees. The main challenge is to find a phylogenetic tree representing optimal evolutionary ancestry between a set of species or taxa. Computational phylogenetics focuses on the algorithms involved in finding optimal phylogenetic tree in the phylogenetic landscape.
Molecular phylogenetics is the branch of phylogeny that analyzes genetic, hereditary molecular differences, predominantly in DNA sequences, to gain information on an organism's evolutionary relationships. From these analyses, it is possible to determine the processes by which diversity among species has been achieved. The result of a molecular phylogenetic analysis is expressed in a phylogenetic tree. Molecular phylogenetics is one aspect of molecular systematics, a broader term that also includes the use of molecular data in taxonomy and biogeography.

Phylogenesis is the biological process by which a taxon appears. The science that studies these processes is called phylogenetics.
Evolutionary taxonomy, evolutionary systematics or Darwinian classification is a branch of biological classification that seeks to classify organisms using a combination of phylogenetic relationship, progenitor-descendant relationship, and degree of evolutionary change. This type of taxonomy may consider whole taxa rather than single species, so that groups of species can be inferred as giving rise to new groups. The concept found its most well-known form in the modern evolutionary synthesis of the early 1940s.

In cladistics or phylogenetics, an outgroup is a more distantly related group of organisms that serves as a reference group when determining the evolutionary relationships of the ingroup, the set of organisms under study, and is distinct from sociological outgroups. The outgroup is used as a point of comparison for the ingroup and specifically allows for the phylogeny to be rooted. Because the polarity (direction) of character change can be determined only on a rooted phylogeny, the choice of outgroup is essential for understanding the evolution of traits along a phylogeny.
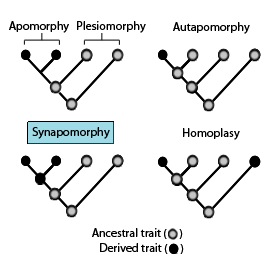
In phylogenetics, an apomorphy is a novel character or character state that has evolved from its ancestral form. A synapomorphy is an apomorphy shared by two or more taxa and is therefore hypothesized to have evolved in their most recent common ancestor. In cladistics, synapomorphy implies homology.
In phylogenetics and computational phylogenetics, maximum parsimony is an optimality criterion under which the phylogenetic tree that minimizes the total number of character-state changes. Under the maximum-parsimony criterion, the optimal tree will minimize the amount of homoplasy. In other words, under this criterion, the shortest possible tree that explains the data is considered best. Some of the basic ideas behind maximum parsimony were presented by James S. Farris in 1970 and Walter M. Fitch in 1971.
In phylogenetics, long branch attraction (LBA) is a form of systematic error whereby distantly related lineages are incorrectly inferred to be closely related. LBA arises when the amount of molecular or morphological change accumulated within a lineage is sufficient to cause that lineage to appear similar to another long-branched lineage, solely because they have both undergone a large amount of change, rather than because they are related by descent. Such bias is more common when the overall divergence of some taxa results in long branches within a phylogeny. Long branches are often attracted to the base of a phylogenetic tree, because the lineage included to represent an outgroup is often also long-branched. The frequency of true LBA is unclear and often debated, and some authors view it as untestable and therefore irrelevant to empirical phylogenetic inference. Although often viewed as a failing of parsimony-based methodology, LBA could in principle result from a variety of scenarios and be inferred under multiple analytical paradigms.
In phylogenetics, a primitive character, trait, or feature of a lineage or taxon is one that is inherited from the common ancestor of a clade and has undergone little change since. Conversely, a trait that appears within the clade group is called advanced or derived. A clade is a group of organisms that consists of a common ancestor and all its lineal descendants.
Computational phylogenetics, phylogeny inference, or phylogenetic inference focuses on computational and optimization algorithms, heuristics, and approaches involved in phylogenetic analyses. The goal is to find a phylogenetic tree representing optimal evolutionary ancestry between a set of genes, species, or taxa. Maximum likelihood, parsimony, Bayesian, and minimum evolution are typical optimality criteria used to assess how well a phylogenetic tree topology describes the sequence data. Nearest Neighbour Interchange (NNI), Subtree Prune and Regraft (SPR), and Tree Bisection and Reconnection (TBR), known as tree rearrangements, are deterministic algorithms to search for optimal or the best phylogenetic tree. The space and the landscape of searching for the optimal phylogenetic tree is known as phylogeny search space.
Ancestral reconstruction is the extrapolation back in time from measured characteristics of individuals to their common ancestors. It is an important application of phylogenetics, the reconstruction and study of the evolutionary relationships among individuals, populations or species to their ancestors. In the context of evolutionary biology, ancestral reconstruction can be used to recover different kinds of ancestral character states of organisms that lived millions of years ago. These states include the genetic sequence, the amino acid sequence of a protein, the composition of a genome, a measurable characteristic of an organism (phenotype), and the geographic range of an ancestral population or species. This is desirable because it allows us to examine parts of phylogenetic trees corresponding to the distant past, clarifying the evolutionary history of the species in the tree. Since modern genetic sequences are essentially a variation of ancient ones, access to ancient sequences may identify other variations and organisms which could have arisen from those sequences. In addition to genetic sequences, one might attempt to track the changing of one character trait to another, such as fins turning to legs.
Distance matrices are used in phylogeny as non-parametric distance methods and were originally applied to phenetic data using a matrix of pairwise distances. These distances are then reconciled to produce a tree. The distance matrix can come from a number of different sources, including measured distance or morphometric analysis, various pairwise distance formulae applied to discrete morphological characters, or genetic distance from sequence, restriction fragment, or allozyme data. For phylogenetic character data, raw distance values can be calculated by simply counting the number of pairwise differences in character states.
Implied weighting describes a group of methods used in phylogenetic analysis to assign the greatest importance to characters that are most likely to be homologous. These are a posteriori methods, which include also dynamic weighting, as opposed to a priori methods, which include adaptive, independent, and chemical categories.

The following outline is provided as an overview of and topical guide to evolution:
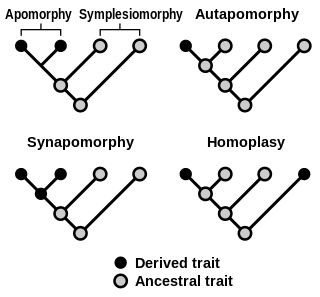
Homoplasy, in biology and phylogenetics, is the term used to describe a feature that has been gained or lost independently in separate lineages over the course of evolution. This is different from homology, which is the term used to characterize the similarity of features that can be parsimoniously explained by common ancestry. Homoplasy can arise from both similar selection pressures acting on adapting species, and the effects of genetic drift.
This glossary of genetics and evolutionary biology is a list of definitions of terms and concepts used in the study of genetics and evolutionary biology, as well as sub-disciplines and related fields, with an emphasis on classical genetics, quantitative genetics, population biology, phylogenetics, speciation, and systematics. Overlapping and related terms can be found in Glossary of cellular and molecular biology, Glossary of ecology, and Glossary of biology.