
Charcot–Marie–Tooth disease (CMT) is a hereditary motor and sensory neuropathy of the peripheral nervous system characterized by progressive loss of muscle tissue and touch sensation across various parts of the body. This disease is the most commonly inherited neurological disorder, affecting about one in 2,500 people. It is named after those who classically described it: the Frenchman Jean-Martin Charcot (1825–1893), his pupil Pierre Marie (1853–1940), and the Briton Howard Henry Tooth (1856–1925).

Spinal muscular atrophies (SMAs) are a genetically and clinically heterogeneous group of rare debilitating disorders characterised by the degeneration of lower motor neurons and subsequent atrophy (wasting) of various muscle groups in the body. While some SMAs lead to early infant death, other diseases of this group permit normal adult life with only mild weakness.
Muscular Dystrophy Association (MDA) is an American nonprofit organization dedicated to supporting people living with muscular dystrophy, ALS, and related neuromuscular diseases. Founded in 1950 by Paul Cohen, who lived with muscular dystrophy, MDA accelerates research, advances care, and works to empower families to live longer and more independent lives but is perhaps known for its working relationship with world-renowned comedian, actor and entertainer Jerry Lewis, its national chairman of 55 years and host of the annual telethon held each Labor Day live on-air. The organization's headquarters is in Chicago, Illinois.
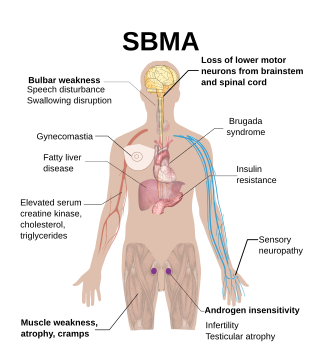
Spinal and bulbar muscular atrophy (SBMA), popularly known as Kennedy's disease, is a rare, adult-onset, X-linked recessive lower motor neuron disease caused by trinucleotide CAG repeat expansions in exon 1 of the androgen receptor (AR) gene, which results in both loss of AR function and toxic gain of function.

A neuromuscular disease is any disease affecting the peripheral nervous system (PNS), the neuromuscular junctions, or skeletal muscles, all of which are components of the motor unit. Damage to any of these structures can cause muscle atrophy and weakness. Issues with sensation can also occur.
Neuromuscular medicine is a subspecialty of neurology and physiatry that focuses the diagnosis and management of neuromuscular diseases. The field encompasses issues related to both diagnosis and management of these conditions, including rehabilitation interventions to optimize the quality of life of individuals with these conditions. This field encompasses disorders that impact both adults and children and which can be inherited or acquired, typically from an autoimmune disease. A neurologist or physiatrist can diagnose these diseases through a clinical history, examination, and electromyography including nerve conduction studies. Many recent drug therapies have been developed to address the acquired neuromuscular diseases including but not limited to immune suppression and drugs that increase the neurotransmitters at the neuromuscular junction. Gene modifying therapies are also a recent treatment branch of neuromuscular medicine with advancements made in disorders such as spinal muscular atrophy and Duchenne muscular dystrophy.

Progressive muscular atrophy (PMA), also called Duchenne–Aran disease and Duchenne–Aran muscular atrophy, is a disorder characterised by the degeneration of lower motor neurons, resulting in generalised, progressive loss of muscle function.
Spinal muscular atrophy (SMA) is a rare neuromuscular disorder that results in the loss of motor neurons and progressive muscle wasting. It is usually diagnosed in infancy or early childhood and if left untreated it is the most common genetic cause of infant death. It may also appear later in life and then have a milder course of the disease. The common feature is progressive weakness of voluntary muscles, with arm, leg and respiratory muscles being affected first. Associated problems may include poor head control, difficulties swallowing, scoliosis, and joint contractures.

Glycine—tRNA ligase also known as glycyl–tRNA synthetase is an enzyme that in humans is encoded by the GARS1 gene.

Hereditary motor and sensory neuropathies (HMSN) is a name sometimes given to a group of different neuropathies which are all characterized by their impact upon both afferent and efferent neural communication. HMSN are characterised by atypical neural development and degradation of neural tissue. The two common forms of HMSN are either hypertrophic demyelinated nerves or complete atrophy of neural tissue. Hypertrophic condition causes neural stiffness and a demyelination of nerves in the peripheral nervous system, and atrophy causes the breakdown of axons and neural cell bodies. In these disorders, a patient experiences progressive muscle atrophy and sensory neuropathy of the extremities.

X-linked spinal muscular atrophy type 2, also known as arthrogryposis multiplex congenita X-linked type 1 (AMCX1), is a rare neurological disorder involving death of motor neurons in the anterior horn of spinal cord resulting in generalised muscle wasting (atrophy). The disease is caused by a mutation in UBA1 gene and is passed in an X-linked recessive manner by carrier mothers to affected sons.

Distal spinal muscular atrophy type 1 (DSMA1), also known as spinal muscular atrophy with respiratory distress type 1 (SMARD1), is a rare neuromuscular disorder involving death of motor neurons in the spinal cord which leads to a generalised progressive atrophy of body muscles.

Olesoxime (TRO19622) is an experimental drug formerly under development by the now-defunct French company Trophos as a treatment for a range of neuromuscular disorders. It has a cholesterol-like structure and belongs to the cholesterol-oxime family of mitochondrial pore modulators.

Spinal muscular atrophy with lower extremity predominance 1 (SMALED1) is an extremely rare neuromuscular disorder of infants characterised by severe progressive muscle atrophy which is especially prominent in legs.
Spinal muscular atrophy with progressive myoclonic epilepsy (SMA-PME), sometimes called Jankovic–Rivera syndrome, is a very rare neurodegenerative disease whose symptoms include slowly progressive muscle loss (atrophy), predominantly affecting proximal muscles, combined with denervation and myoclonic seizures. Only 12 known human families are described in scientific literature to have SMA-PME.

Nusinersen, marketed as Spinraza, is a medication used in treating spinal muscular atrophy (SMA), a rare neuromuscular disorder. In December 2016, it became the first approved drug used in treating this disorder.
Distal hereditary motor neuropathy type V is a particular type of neuropathic disorder. In general, distal hereditary motor neuropathies affect the axons of distal motor neurons and are characterized by progressive weakness and atrophy of muscles of the extremities. It is common for them to be called "spinal forms of Charcot-Marie-Tooth disease (CMT)", because the diseases are closely related in symptoms and genetic cause. The diagnostic difference in these diseases is the presence of sensory loss in the extremities. There are seven classifications of dHMNs, each defined by patterns of inheritance, age of onset, severity, and muscle groups involved. Type V is a disorder characterized by autosomal dominance, weakness of the upper limbs that is progressive and symmetrical, and atrophy of the small muscles of the hands.

Risdiplam, sold under the brand name Evrysdi, is a medication used to treat spinal muscular atrophy (SMA) and the first oral medication approved to treat this disease.

Michael Jeffrey Aminoff is a clinical neurologist and neurophysiologist whose later clinical work focused on treating Parkinson's disease and related movement disorders. He retired in 2022 and lives in San Francisco, California.
Mary M. Reilly FRCP is an Irish neurologist who works at National Hospital for Neurology and Neurosurgery. She studies peripheral neuropathy. She is the President of the Association of British Neurologists.
