
Phenylketonuria (PKU) is an inborn error of metabolism that results in decreased metabolism of the amino acid phenylalanine. Untreated PKU can lead to intellectual disability, seizures, behavioral problems, and mental disorders. It may also result in a musty smell and lighter skin. A baby born to a mother who has poorly treated PKU may have heart problems, a small head, and low birth weight.

Clinical chemistry is a division in medical laboratory sciences focusing on qualitative tests of important compounds, referred to as analytes or markers, in bodily fluids and tissues using analytical techniques and specialized instruments. This interdisciplinary field includes knowledge from medicine, biology, chemistry, biomedical engineering, informatics, and an applied form of biochemistry.

The neonatal heel prick is a blood collection procedure done on newborns. It consists of making a pinprick puncture in one heel of the newborn to collect their blood. This technique is used frequently as the main way to collect blood from neonates. Other techniques include venous or arterial needle sticks, cord blood sampling, or umbilical line collection. This technique is often utilized for the Guthrie test, where it is used to soak the blood into pre-printed collection cards known as Guthrie cards.

The Université de Sherbrooke is a French-language public research university in Sherbrooke, Quebec, Canada, with a second campus in Longueuil, a suburb on the South Shore of Montreal. It is one of two universities in the Estrie region of Quebec, and the only French-language university for the region.

Medium-chain acyl-CoA dehydrogenase deficiency is a disorder of fatty acid oxidation that impairs the body's ability to break down medium-chain fatty acids into acetyl-CoA. The disorder is characterized by hypoglycemia and sudden death without timely intervention, most often brought on by periods of fasting or vomiting.
Newborn screening (NBS) is a public health program of screening in infants shortly after birth for conditions that are treatable, but not clinically evident in the newborn period. The goal is to identify infants at risk for these conditions early enough to confirm the diagnosis and provide intervention that will alter the clinical course of the disease and prevent or ameliorate the clinical manifestations. NBS started with the discovery that the amino acid disorder phenylketonuria (PKU) could be treated by dietary adjustment, and that early intervention was required for the best outcome. Infants with PKU appear normal at birth, but are unable to metabolize the essential amino acid phenylalanine, resulting in irreversible intellectual disability. In the 1960s, Robert Guthrie developed a simple method using a bacterial inhibition assay that could detect high levels of phenylalanine in blood shortly after a baby was born. Guthrie also pioneered the collection of blood on filter paper which could be easily transported, recognizing the need for a simple system if the screening was going to be done on a large scale. Newborn screening around the world is still done using similar filter paper. NBS was first introduced as a public health program in the United States in the early 1960s, and has expanded to countries around the world.
Inborn errors of metabolism form a large class of genetic diseases involving congenital disorders of enzyme activities. The majority are due to defects of single genes that code for enzymes that facilitate conversion of various substances (substrates) into others (products). In most of the disorders, problems arise due to accumulation of substances which are toxic or interfere with normal function, or due to the effects of reduced ability to synthesize essential compounds. Inborn errors of metabolism are often referred to as congenital metabolic diseases or inherited metabolic disorders. Another term used to describe these disorders is "enzymopathies". This term was created following the study of biodynamic enzymology, a science based on the study of the enzymes and their products. Finally, inborn errors of metabolism were studied for the first time by British physician Archibald Garrod (1857–1936), in 1908. He is known for work that prefigured the "one gene-one enzyme" hypothesis, based on his studies on the nature and inheritance of alkaptonuria. His seminal text, Inborn Errors of Metabolism, was published in 1923.

Isovaleric acidemia is a rare autosomal recessive metabolic disorder which disrupts or prevents normal metabolism of the branched-chain amino acid leucine. It is a classical type of organic acidemia.

3-Hydroxy-3-methylglutaryl-CoA lyase deficiency, (HMGCLD) also known as HMGCL deficiency, HMG-CoA lyase deficiency, or hydroxymethylglutaric aciduria, is an uncommon autosomal recessive inborn error in ketone body production and leucine breakdown caused by HMGCL gene mutations. HMGCL, located on chromosome 1p36.11's short arm, codes for HMG-CoA lyase, which aids in the metabolism of dietary proteins by converting HMG-CoA into acetyl-CoA and acetoacetate.

Systemic primary carnitine deficiency (SPCD) is an inborn error of fatty acid transport caused by a defect in the transporter responsible for moving carnitine across the plasma membrane. Carnitine is an important amino acid for fatty acid metabolism. When carnitine cannot be transported into tissues, fatty acid oxidation is impaired, leading to a variety of symptoms such as chronic muscle weakness, cardiomyopathy, hypoglycemia and liver dysfunction. The specific transporter involved with SPCD is OCTN2, coded for by the SLC22A5 gene located on chromosome 5. SPCD is inherited in an autosomal recessive manner, with mutated alleles coming from both parents.

Professor Michael H. Gleb is an American biochemist and chemist specializing in enzymes and particularly those of medical significance. He is the Boris and Barbara L. Weinstein Endowed Chair in Chemistry at the University of Washington in Seattle. He also teaches Honors Organic Chemistry, Chemical Biology and Enzymology.
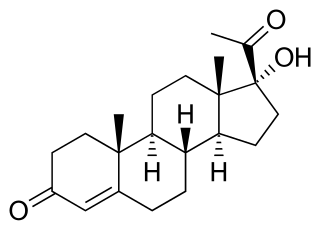
17α-Hydroxyprogesterone (17α-OHP), also known as 17-OH progesterone (17-OHP), or hydroxyprogesterone (OHP), is an endogenous progestogen steroid hormone related to progesterone. It is also a chemical intermediate in the biosynthesis of many other endogenous steroids, including androgens, estrogens, glucocorticoids, and mineralocorticoids, as well as neurosteroids.

Group B streptococcal infection, also known as Group B streptococcal disease or just Group B strep infection, is the infectious disease caused by the bacterium Streptococcus agalactiae, which is the most common human pathogen belonging to the group B of the Lancefield classification of streptococci—hence the group B stretococcal (GBS) infection nomenclature. Infection with GBS can cause serious illness and sometimes death, especially in newborns, the elderly, and people with compromised immune systems. The most severe form of group B streptococcal disease is neonatal meningitis in infants, which is frequently lethal and can cause permanent neuro-cognitive impairment.

Argininosuccinic aciduria is an inherited disorder that causes the accumulation of argininosuccinic acid in the blood and urine. Some patients may also have an elevation of ammonia, a toxic chemical, which can affect the nervous system. Argininosuccinic aciduria may become evident in the first few days of life because of high blood ammonia, or later in life presenting with "sparse" or "brittle" hair, developmental delay, and tremors.

3-Methylcrotonyl-CoA carboxylase deficiency also known as 3-Methylcrotonylglycinuria is an inborn error of leucine metabolism and is inherited through an autosomal recessive fashion. 3-Methylcrotonyl-CoA carboxylase deficiency is caused by mutations in the MCCC1 gene, formerly known as MMCA, or the MCCC2 gene, formerly known as MCCB. MCCC1 encodes the a-subunits of 3-methylcrotonyl-CoA carboxylase while MCCC2 encodes the b-subunits. The clinical presentation of 3-Methylcrotonyl-CoA carboxylase deficiency is varied, even within members of the same family.

Histidinemia is a rare autosomal recessive metabolic disorder caused by a deficiency of the enzyme histidase. Histidase is needed for the metabolism of the amino acid histidine. Although originally thought to be linked to multiple developmental disorders histidinemia is now accepted as a relatively benign disorder, leading to a reduction in the prevalence of neonatal screening procedures.

Guanidinoacetate methyltransferase deficiency is an autosomal recessive cerebral creatine deficiency that primarily affects the nervous system and muscles. It is the first described disorder of creatine metabolism, and results from deficient activity of guanidinoacetate methyltransferase, an enzyme involved in the synthesis of creatine. Clinically, affected individuals often present with hypotonia, seizures and developmental delay. Diagnosis can be suspected on clinical findings, and confirmed by specific biochemical tests, brain magnetic resonance spectroscopy, or genetic testing. Biallelic pathogenic variants in GAMT are the underlying cause of the disorder. After GAMT deficiency is diagnosed, it can be treated by dietary adjustments, including supplementation with creatine. Treatment is highly effective if started early in life. If treatment is started late, it cannot reverse brain damage which has already taken place.
Organic acidemia is a term used to classify a group of metabolic disorders which disrupt normal amino acid metabolism, particularly branched-chain amino acids, causing a buildup of acids which are usually not present.
Combined malonic and methylmalonic aciduria (CMAMMA), also called combined malonic and methylmalonic acidemia is an inherited metabolic disease characterized by elevated levels of malonic acid and methylmalonic acid. However, the methylmalonic acid levels exceed those of malonic acid. CMAMMA is not only an organic aciduria but also a defect of mitochondrial fatty acid synthesis (mtFASII). Some researchers have hypothesized that CMAMMA might be one of the most common forms of methylmalonic acidemia, and possibly one of the most common inborn errors of metabolism. Due to being infrequently diagnosed, it most often goes undetected.

Claude Laberge, C.M., O.Q., M.D., Ph.D., FRCP(C) is a physician-geneticist and a Professor Emeritus of Medicine and Pediatrics at the Faculty of Medicine at Université Laval in Québec City, Québec. He is a pioneer in the field of human genetics.