Related Research Articles

A DNA sequencer is a scientific instrument used to automate the DNA sequencing process. Given a sample of DNA, a DNA sequencer is used to determine the order of the four bases: G (guanine), C (cytosine), A (adenine) and T (thymine). This is then reported as a text string, called a read. Some DNA sequencers can be also considered optical instruments as they analyze light signals originating from fluorochromes attached to nucleotides.
In bioinformatics, sequence assembly refers to aligning and merging fragments from a longer DNA sequence in order to reconstruct the original sequence. This is needed as DNA sequencing technology might not be able to 'read' whole genomes in one go, but rather reads small pieces of between 20 and 30,000 bases, depending on the technology used. Typically, the short fragments (reads) result from shotgun sequencing genomic DNA, or gene transcript (ESTs).

BioPerl is a collection of Perl modules that facilitate the development of Perl scripts for bioinformatics applications. It has played an integral role in the Human Genome Project.
Pyrosequencing is a method of DNA sequencing based on the "sequencing by synthesis" principle, in which the sequencing is performed by detecting the nucleotide incorporated by a DNA polymerase. Pyrosequencing relies on light detection based on a chain reaction when pyrophosphate is released. Hence, the name pyrosequencing.

DNA sequencing is the process of determining the nucleic acid sequence – the order of nucleotides in DNA. It includes any method or technology that is used to determine the order of the four bases: adenine, guanine, cytosine, and thymine. The advent of rapid DNA sequencing methods has greatly accelerated biological and medical research and discovery.
Metagenomics is the study of genetic material recovered directly from environmental or clinical samples by a method called sequencing. The broad field may also be referred to as environmental genomics, ecogenomics, community genomics or microbiomics.

Sanger sequencing is a method of DNA sequencing that involves electrophoresis and is based on the random incorporation of chain-terminating dideoxynucleotides by DNA polymerase during in vitro DNA replication. After first being developed by Frederick Sanger and colleagues in 1977, it became the most widely used sequencing method for approximately 40 years. It was first commercialized by Applied Biosystems in 1986. More recently, higher volume Sanger sequencing has been replaced by next generation sequencing methods, especially for large-scale, automated genome analyses. However, the Sanger method remains in wide use for smaller-scale projects and for validation of deep sequencing results. It still has the advantage over short-read sequencing technologies in that it can produce DNA sequence reads of > 500 nucleotides and maintains a very low error rate with accuracies around 99.99%. Sanger sequencing is still actively being used in efforts for public health initiatives such as sequencing the spike protein from SARS-CoV-2 as well as for the surveillance of norovirus outbreaks through the Center for Disease Control and Prevention's (CDC) CaliciNet surveillance network.

William James Kent is an American research scientist and computer programmer. He has been a contributor to genome database projects and the 2003 winner of the Benjamin Franklin Award.
The Human Genome Project (HGP) was an international scientific research project with the goal of determining the base pairs that make up human DNA, and of identifying, mapping and sequencing all of the genes of the human genome from both a physical and a functional standpoint. It started in 1990 and was completed in 2006. It remains the world's largest collaborative biological project. Planning for the project started after it was adopted in 1984 by the US government, and it officially launched in 1990. It was declared complete on April 14, 2003, and included about 92% of the genome. Level "complete genome" was achieved in May 2021, with a remaining only 0.3% bases covered by potential issues. The final gapless assembly was finished in January 2022.

A Phred quality score is a measure of the quality of the identification of the nucleobases generated by automated DNA sequencing. It was originally developed for the computer program Phred to help in the automation of DNA sequencing in the Human Genome Project. Phred quality scores are assigned to each nucleotide base call in automated sequencer traces. The FASTQ format encodes phred scores as ASCII characters alongside the read sequences. Phred quality scores have become widely accepted to characterize the quality of DNA sequences, and can be used to compare the efficacy of different sequencing methods. Perhaps the most important use of Phred quality scores is the automatic determination of accurate, quality-based consensus sequences.
The Baylor College of Medicine Human Genome Sequencing Center (BCM-HGSC) was established by Richard A. Gibbs in 1996 when Baylor College of Medicine was chosen as one of six worldwide sites to complete the final phase of the international Human Genome Project. Gibbs is the current director of the BCM-HGSC.

Bardet–Biedl syndrome 5 protein is a protein that in humans is encoded by the BBS5 gene.
Phred is a computer program for base calling, that is to say, identifying a nucleobase sequence from fluorescence "trace" data generated by an automated DNA sequencer that uses electrophoresis and 4-fluorescent dye method. When originally developed, Phred produced significantly fewer errors in the data sets examined than other methods, averaging 40–50% fewer errors. Phred quality scores have become widely accepted to characterize the quality of DNA sequences, and can be used to compare the efficacy of different sequencing methods.
Phrap is a widely used program for DNA sequence assembly. It is part of the Phred-Phrap-Consed package.
The Staden Package is computer software, a set of tools for DNA sequence assembly, editing, and sequence analysis. It is open-source software, released under a BSD 3-clause license.
In molecular biology, and more importantly high-throughput DNA sequencing, a chimera is a single DNA sequence originating when multiple transcripts or DNA sequences get joined. Chimeras can be considered artifacts and be filtered out from the data during processing to prevent spurious inferences of biological variation. However, chimeras should not be confused with chimeric reads, who are generally used by structural variant callers to detect structural variation events and are not always an indication of the presence of a chimeric transcript or gene.
SOAP is a suite of bioinformatics software tools from the BGI Bioinformatics department enabling the assembly, alignment, and analysis of next generation DNA sequencing data. It is particularly suited to short read sequencing data.

A reference genome is a digital nucleic acid sequence database, assembled by scientists as a representative example of the set of genes in one idealized individual organism of a species. As they are assembled from the sequencing of DNA from a number of individual donors, reference genomes do not accurately represent the set of genes of any single individual organism. Instead, a reference provides a haploid mosaic of different DNA sequences from each donor. For example, one of the most recent human reference genomes, assembly GRCh38/hg38, is derived from >60 genomic clone libraries. There are reference genomes for multiple species of viruses, bacteria, fungus, plants, and animals. Reference genomes are typically used as a guide on which new genomes are built, enabling them to be assembled much more quickly and cheaply than the initial Human Genome Project. Reference genomes can be accessed online at several locations, using dedicated browsers such as Ensembl or UCSC Genome Browser.
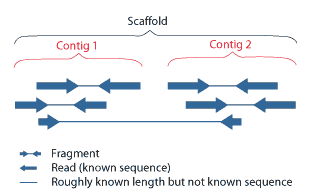
Scaffolding is a technique used in bioinformatics. It is defined as follows:
Link together a non-contiguous series of genomic sequences into a scaffold, consisting of sequences separated by gaps of known length. The sequences that are linked are typically contiguous sequences corresponding to read overlaps.
De novo sequence assemblers are a type of program that assembles short nucleotide sequences into longer ones without the use of a reference genome. These are most commonly used in bioinformatic studies to assemble genomes or transcriptomes. Two common types of de novo assemblers are greedy algorithm assemblers and De Bruijn graph assemblers.
References
- ↑ Gordon D, Abajian C, Green P (1998). "Consed: A Graphical Tool for Sequence Finishing". Genome Research. 8 (3): 195–202. doi: 10.1101/gr.8.3.195 . PMID 9521923.
- ↑ Gordon D, Desmarais C, Green P (2001). "Automated Finishing with Autofinish". Genome Research. 11 (4): 614–625. doi:10.1101/gr.171401. PMC 311035 . PMID 11282977.