Bra–ket notation, also called Dirac notation, is a notation for linear algebra and linear operators on complex vector spaces together with their dual space both in the finite-dimensional and infinite-dimensional case. It is specifically designed to ease the types of calculations that frequently come up in quantum mechanics. Its use in quantum mechanics is quite widespread.
A hydrogen atom is an atom of the chemical element hydrogen. The electrically neutral atom contains a single positively charged proton and a single negatively charged electron bound to the nucleus by the Coulomb force. Atomic hydrogen constitutes about 75% of the baryonic mass of the universe.
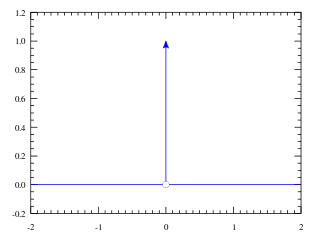
In mathematical analysis, the Dirac delta function, also known as the unit impulse, is a generalized function on the real numbers, whose value is zero everywhere except at zero, and whose integral over the entire real line is equal to one. Since there is no function having this property, to model the delta "function" rigorously involves the use of limits or, as is common in mathematics, measure theory and the theory of distributions.
Electron density or electronic density is the measure of the probability of an electron being present at an infinitesimal element of space surrounding any given point. It is a scalar quantity depending upon three spatial variables and is typically denoted as either or . The density is determined, through definition, by the normalised -electron wavefunction which itself depends upon variables. Conversely, the density determines the wave function modulo up to a phase factor, providing the formal foundation of density functional theory.
Coupled cluster (CC) is a numerical technique used for describing many-body systems. Its most common use is as one of several post-Hartree–Fock ab initio quantum chemistry methods in the field of computational chemistry, but it is also used in nuclear physics. Coupled cluster essentially takes the basic Hartree–Fock molecular orbital method and constructs multi-electron wavefunctions using the exponential cluster operator to account for electron correlation. Some of the most accurate calculations for small to medium-sized molecules use this method.

The path integral formulation is a description in quantum mechanics that generalizes the stationary action principle of classical mechanics. It replaces the classical notion of a single, unique classical trajectory for a system with a sum, or functional integral, over an infinity of quantum-mechanically possible trajectories to compute a quantum amplitude.
In computational physics and chemistry, the Hartree–Fock (HF) method is a method of approximation for the determination of the wave function and the energy of a quantum many-body system in a stationary state.
In the Hartree–Fock method of quantum mechanics, the Fock matrix is a matrix approximating the single-electron energy operator of a given quantum system in a given set of basis vectors. It is most often formed in computational chemistry when attempting to solve the Roothaan equations for an atomic or molecular system. The Fock matrix is actually an approximation to the true Hamiltonian operator of the quantum system. It includes the effects of electron-electron repulsion only in an average way. Because the Fock operator is a one-electron operator, it does not include the electron correlation energy.
Møller–Plesset perturbation theory (MP) is one of several quantum chemistry post-Hartree–Fock ab initio methods in the field of computational chemistry. It improves on the Hartree–Fock method by adding electron correlation effects by means of Rayleigh–Schrödinger perturbation theory (RS-PT), usually to second (MP2), third (MP3) or fourth (MP4) order. Its main idea was published as early as 1934 by Christian Møller and Milton S. Plesset.
Multi-configurational self-consistent field (MCSCF) is a method in quantum chemistry used to generate qualitatively correct reference states of molecules in cases where Hartree–Fock and density functional theory are not adequate. It uses a linear combination of configuration state functions (CSF), or configuration determinants, to approximate the exact electronic wavefunction of an atom or molecule. In an MCSCF calculation, the set of coefficients of both the CSFs or determinants and the basis functions in the molecular orbitals are varied to obtain the total electronic wavefunction with the lowest possible energy. This method can be considered a combination between configuration interaction and Hartree–Fock.
In quantum mechanics, the exchange operator, also known as permutation operator, is a quantum mechanical operator that acts on states in Fock space. The exchange operator acts by switching the labels on any two identical particles described by the joint position quantum state . Since the particles are identical, the notion of exchange symmetry requires that the exchange operator be unitary.
The Kohn-Sham equations are a set of mathematical equations used in quantum mechanics to simplify the complex problem of understanding how electrons behave in atoms and molecules. They introduce fictitious non-interacting electrons and use them to find the most stable arrangement of electrons, which helps scientists understand and predict the properties of matter at the atomic and molecular scale.
In atomic, molecular, and optical physics and quantum chemistry, the molecular Hamiltonian is the Hamiltonian operator representing the energy of the electrons and nuclei in a molecule. This operator and the associated Schrödinger equation play a central role in computational chemistry and physics for computing properties of molecules and aggregates of molecules, such as thermal conductivity, specific heat, electrical conductivity, optical, and magnetic properties, and reactivity.
Within computational chemistry, the Slater–Condon rules express integrals of one- and two-body operators over wavefunctions constructed as Slater determinants of orthonormal orbitals in terms of the individual orbitals. In doing so, the original integrals involving N-electron wavefunctions are reduced to sums over integrals involving at most two molecular orbitals, or in other words, the original 3N dimensional integral is expressed in terms of many three- and six-dimensional integrals.
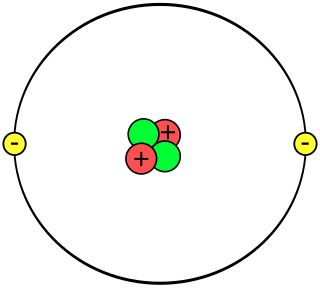
A helium atom is an atom of the chemical element helium. Helium is composed of two electrons bound by the electromagnetic force to a nucleus containing two protons along with two neutrons, depending on the isotope, held together by the strong force. Unlike for hydrogen, a closed-form solution to the Schrödinger equation for the helium atom has not been found. However, various approximations, such as the Hartree–Fock method, can be used to estimate the ground state energy and wavefunction of the atom. Historically, the first such helium spectrum calculation was done by Albrecht Unsöld in 1927. Its success was considered to be one of the earliest signs of validity of Schrödinger's wave mechanics.
In quantum chemistry, Brillouin's theorem, proposed by the French physicist Léon Brillouin in 1934, relates to Hartree–Fock wavefunctions. Hartree–Fock, or the self-consistent field method, is a non-relativistic method of generating approximate wavefunctions for a many-bodied quantum system, based on the assumption that each electron is exposed to an average of the positions of all other electrons, and that the solution is a linear combination of pre-specified basis functions.
Static force fields are fields, such as a simple electric, magnetic or gravitational fields, that exist without excitations. The most common approximation method that physicists use for scattering calculations can be interpreted as static forces arising from the interactions between two bodies mediated by virtual particles, particles that exist for only a short time determined by the uncertainty principle. The virtual particles, also known as force carriers, are bosons, with different bosons associated with each force.
The Wannier equation describes a quantum mechanical eigenvalue problem in solids where an electron in a conduction band and an electronic vacancy within a valence band attract each other via the Coulomb interaction. For one electron and one hole, this problem is analogous to the Schrödinger equation of the hydrogen atom; and the bound-state solutions are called excitons. When an exciton's radius extends over several unit cells, it is referred to as a Wannier exciton in contrast to Frenkel excitons whose size is comparable with the unit cell. An excited solid typically contains many electrons and holes; this modifies the Wannier equation considerably. The resulting generalized Wannier equation can be determined from the homogeneous part of the semiconductor Bloch equations or the semiconductor luminescence equations.
Symmetries in quantum mechanics describe features of spacetime and particles which are unchanged under some transformation, in the context of quantum mechanics, relativistic quantum mechanics and quantum field theory, and with applications in the mathematical formulation of the standard model and condensed matter physics. In general, symmetry in physics, invariance, and conservation laws, are fundamentally important constraints for formulating physical theories and models. In practice, they are powerful methods for solving problems and predicting what can happen. While conservation laws do not always give the answer to the problem directly, they form the correct constraints and the first steps to solving a multitude of problems.
In quantum probability, the Belavkin equation, also known as Belavkin-Schrödinger equation, quantum filtering equation, stochastic master equation, is a quantum stochastic differential equation describing the dynamics of a quantum system undergoing observation in continuous time. It was derived and henceforth studied by Viacheslav Belavkin in 1988.








