Related Research Articles

Cardiomyopathy is a group of diseases that affect the heart muscle. Early on there may be few or no symptoms. As the disease worsens, shortness of breath, feeling tired, and swelling of the legs may occur, due to the onset of heart failure. An irregular heart beat and fainting may occur. Those affected are at an increased risk of sudden cardiac death.
Hypertrophic cardiomyopathy is a condition in which the heart becomes thickened without an obvious cause. The parts of the heart most commonly affected are the interventricular septum and the ventricles. This results in the heart being less able to pump blood effectively and also may cause electrical conduction problems.

Desmin is a protein that in humans is encoded by the DES gene. Desmin is a muscle-specific, type III intermediate filament that integrates the sarcolemma, Z disk, and nuclear membrane in sarcomeres and regulates sarcomere architecture.
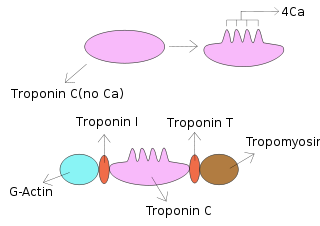
Troponin C is a protein which is part of the troponin complex. It contains four calcium-binding EF hands, although different isoforms may have fewer than four functional calcium-binding subdomains. It is a component of thin filaments, along with actin and tropomyosin. It contains an N lobe and a C lobe. The C lobe serves a structural purpose and binds to the N domain of troponin I (TnI). The C lobe can bind either Ca2+ or Mg2+. The N lobe, which binds only Ca2+, is the regulatory lobe and binds to the C domain of troponin I after calcium binding.

Troponin I, cardiac muscle is a protein that in humans is encoded by the TNNI3 gene. It is a tissue-specific subtype of troponin I, which in turn is a part of the troponin complex.

Cardiac muscle troponin T (cTnT) is a protein that in humans is encoded by the TNNT2 gene. Cardiac TnT is the tropomyosin-binding subunit of the troponin complex, which is located on the thin filament of striated muscles and regulates muscle contraction in response to alterations in intracellular calcium ion concentration.

Tropomyosin alpha-1 chain is a protein that in humans is encoded by the TPM1 gene. This gene is a member of the tropomyosin (Tm) family of highly conserved, widely distributed actin-binding proteins involved in the contractile system of striated and smooth muscles and the cytoskeleton of non-muscle cells.

ACTC1 encodes cardiac muscle alpha actin. This isoform differs from the alpha actin that is expressed in skeletal muscle, ACTA1. Alpha cardiac actin is the major protein of the thin filament in cardiac sarcomeres, which are responsible for muscle contraction and generation of force to support the pump function of the heart.

The myosin-binding protein C, cardiac-type is a protein that in humans is encoded by the MYBPC3 gene. This isoform is expressed exclusively in heart muscle during human and mouse development, and is distinct from those expressed in slow skeletal muscle (MYBPC1) and fast skeletal muscle (MYBPC2).

Troponin C, also known as TN-C or TnC, is a protein that resides in the troponin complex on actin thin filaments of striated muscle and is responsible for binding calcium to activate muscle contraction. Troponin C is encoded by the TNNC1 gene in humans for both cardiac and slow skeletal muscle.

β-Tropomyosin, also known as tropomyosin beta chain is a protein that in humans is encoded by the TPM2 gene. β-tropomyosin is striated muscle-specific coiled coil dimer that functions to stabilize actin filaments and regulate muscle contraction.

Telethonin, also known as Tcap, is a protein that in humans is encoded by the TCAP gene. Telethonin is expressed in cardiac and skeletal muscle at Z-discs and functions to regulate sarcomere assembly, T-tubule function and apoptosis. Telethonin has been implicated in several diseases, including limb-girdle muscular dystrophy, hypertrophic cardiomyopathy, dilated cardiomyopathy and idiopathic cardiomyopathy.

Myosin regulatory light chain 2, ventricular/cardiac muscle isoform (MLC-2) also known as the regulatory light chain of myosin (RLC) is a protein that in humans is encoded by the MYL2 gene. This cardiac ventricular RLC isoform is distinct from that expressed in skeletal muscle (MYLPF), smooth muscle (MYL12B) and cardiac atrial muscle (MYL7).

Slow skeletal muscle troponin T (sTnT) is a protein that in humans is encoded by the TNNT1 gene.

Myosin heavy chain, α isoform (MHC-α) is a protein that in humans is encoded by the MYH6 gene. This isoform is distinct from the ventricular/slow myosin heavy chain isoform, MYH7, referred to as MHC-β. MHC-α isoform is expressed predominantly in human cardiac atria, exhibiting only minor expression in human cardiac ventricles. It is the major protein comprising the cardiac muscle thick filament, and functions in cardiac muscle contraction. Mutations in MYH6 have been associated with late-onset hypertrophic cardiomyopathy, atrial septal defects and sick sinus syndrome.

Myosin essential light chain (ELC), ventricular/cardiac isoform is a protein that in humans is encoded by the MYL3 gene. This cardiac ventricular/slow skeletal ELC isoform is distinct from that expressed in fast skeletal muscle (MYL1) and cardiac atrial muscle (MYL4). Ventricular ELC is part of the myosin molecule and is important in modulating cardiac muscle contraction.
Obscurin is a protein that in humans is encoded by the OBSCN gene. Obscurin belongs to the family of giant sarcomeric signaling proteins that includes titin and nebulin. Obscurin is expressed in cardiac and skeletal muscle, and plays a role in the organization of myofibrils during sarcomere assembly. A mutation in the OBSCN gene has been associated with hypertrophic cardiomyopathy and altered obscurin protein properties have been associated with other muscle diseases.
Cysteine and glycine-rich protein 3 also known as cardiac LIM protein (CLP) or muscle LIM protein (MLP) is a protein that in humans is encoded by the CSRP3 gene.

Myopalladin is a protein that in humans is encoded by the MYPN gene. Myopalladin is a muscle protein responsible for tethering proteins at the Z-disc and for communicating between the sarcomere and the nucleus in cardiac and skeletal muscle
A8V is point mutation on Troponin C (cTNC) that leads to a hypertrophic cardiomyopathy. The coordinated cardiac muscle contraction is regulated by the troponin complex on thin filament (troponin C which is calcium binding, troponin T that plays the role with tropomyosin, and troponin I which has an inhibitory action annulating the S1 ATPase activity in the presence of tropomyosin and troponin and absence of Ca2+). This mutation is determined by the change of Alanine to Valine at nucleotide 23 from C to T. Patients with this type of mutation shows thickness on the left ventricle wall of around 18 mm, compared to the normal this thickness would be 12 mm. Also, A8V affects the Ca2+ binding affinity compared to normal genotype and increased sensitivity on force development.
References
- 1 2 Landstrom, Andrew P.; Parvatiyar, Michelle S.; Pinto, Jose R.; Marquardt, Michelle L.; Bos, J. Martijn; Tester, David J.; Ommen, Steve R.; Potter, James D.; Ackerman, Michael J. (August 2008). "Molecular and functional characterization of novel hypertrophic cardiomyopathy susceptibility mutations in TNNC1-encoded troponin C". Journal of Molecular and Cellular Cardiology. 45 (2): 281–288. doi:10.1016/j.yjmcc.2008.05.003. PMC 2627482 . PMID 18572189.
- ↑ Kalyva, Athanasia; Parthenakis, Fragiskos I.; Marketou, Maria E.; Kontaraki, Joanna E.; Vardas, Panos E. (2014-04-01). "Biochemical characterisation of Troponin C mutations causing hypertrophic and dilated cardiomyopathies". Journal of Muscle Research and Cell Motility. 35 (2): 161–178. doi:10.1007/s10974-014-9382-0. ISSN 1573-2657. PMID 24744096. S2CID 1726747.
- ↑ Solaro, R. John; Rosevear, Paul; Kobayashi, Tomoyoshi (2008-04-25). "The unique functions of cardiac troponin I in the control of cardiac muscle contraction and relaxation". Biochemical and Biophysical Research Communications. Ebashi Memorial Issue. 369 (1): 82–87. doi:10.1016/j.bbrc.2007.12.114. ISSN 0006-291X. PMC 2365727 . PMID 18162178.
- ↑ Pinto, Jose Renato; Parvatiyar, Michelle S.; Jones, Michelle A.; Liang, Jingsheng; Ackerman, Michael J.; Potter, James D. (2009-07-10). "A Functional and Structural Study of Troponin C Mutations Related to Hypertrophic Cardiomyopathy". Journal of Biological Chemistry. 284 (28): 19090–19100. doi: 10.1074/jbc.M109.007021 . ISSN 0021-9258. PMC 2707221 . PMID 19439414.
| | This gene article is a stub. You can help Wikipedia by expanding it. |