
Bioinformatics is an interdisciplinary field of science that develops methods and software tools for understanding biological data, especially when the data sets are large and complex. Bioinformatics uses biology, chemistry, physics, computer science, computer programming, information engineering, mathematics and statistics to analyze and interpret biological data. The subsequent process of analyzing and interpreting data is referred to as computational biology.

In the fields of molecular biology and genetics, a genome is all the genetic information of an organism. It consists of nucleotide sequences of DNA. The nuclear genome includes protein-coding genes and non-coding genes, other functional regions of the genome such as regulatory sequences, and often a substantial fraction of junk DNA with no evident function. Almost all eukaryotes have mitochondria and a small mitochondrial genome. Algae and plants also contain chloroplasts with a chloroplast genome.

The human genome is a complete set of nucleic acid sequences for humans, encoded as DNA within the 23 chromosome pairs in cell nuclei and in a small DNA molecule found within individual mitochondria. These are usually treated separately as the nuclear genome and the mitochondrial genome. Human genomes include both protein-coding DNA sequences and various types of DNA that does not encode proteins. The latter is a diverse category that includes DNA coding for non-translated RNA, such as that for ribosomal RNA, transfer RNA, ribozymes, small nuclear RNAs, and several types of regulatory RNAs. It also includes promoters and their associated gene-regulatory elements, DNA playing structural and replicatory roles, such as scaffolding regions, telomeres, centromeres, and origins of replication, plus large numbers of transposable elements, inserted viral DNA, non-functional pseudogenes and simple, highly repetitive sequences. Introns make up a large percentage of non-coding DNA. Some of this non-coding DNA is non-functional junk DNA, such as pseudogenes, but there is no firm consensus on the total amount of junk DNA.

The National Center for Biotechnology Information (NCBI) is part of the United States National Library of Medicine (NLM), a branch of the National Institutes of Health (NIH). It is approved and funded by the government of the United States. The NCBI is located in Bethesda, Maryland, and was founded in 1988 through legislation sponsored by US Congressman Claude Pepper.
Gene duplication is a major mechanism through which new genetic material is generated during molecular evolution. It can be defined as any duplication of a region of DNA that contains a gene. Gene duplications can arise as products of several types of errors in DNA replication and repair machinery as well as through fortuitous capture by selfish genetic elements. Common sources of gene duplications include ectopic recombination, retrotransposition event, aneuploidy, polyploidy, and replication slippage.

In genetics and bioinformatics, a single-nucleotide polymorphism is a germline substitution of a single nucleotide at a specific position in the genome. Although certain definitions require the substitution to be present in a sufficiently large fraction of the population, many publications do not apply such a frequency threshold.
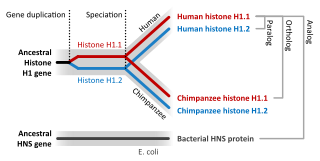
Sequence homology is the biological homology between DNA, RNA, or protein sequences, defined in terms of shared ancestry in the evolutionary history of life. Two segments of DNA can have shared ancestry because of three phenomena: either a speciation event (orthologs), or a duplication event (paralogs), or else a horizontal gene transfer event (xenologs).

Copy number variation (CNV) is a phenomenon in which sections of the genome are repeated and the number of repeats in the genome varies between individuals. Copy number variation is a type of structural variation: specifically, it is a type of duplication or deletion event that affects a considerable number of base pairs. Approximately two-thirds of the entire human genome may be composed of repeats and 4.8–9.5% of the human genome can be classified as copy number variations. In mammals, copy number variations play an important role in generating necessary variation in the population as well as disease phenotype.
The variome is the whole set of genetic variations found in populations of species that have gone through a relatively short evolution change. For example, among humans, about 1 in every 1,200 nucleotide bases differ. The size of human variome in terms of effective population size is claimed to be about 10,000 individuals. This variation rate is comparatively small compared to other species. For example, the effective population size of tigers which perhaps has the whole population size less than 10,000 in the wild is not much smaller than the human species indicating a much higher level of genetic diversity although they are close to extinction in the wild. In practice, the variome can be the sum of the single nucleotide polymorphisms (SNPs), indels, and structural variation (SV) of a population or species. The Human Variome Project seeks to compile this genetic variation data worldwide. Variomics is the study of variome and a branch of bioinformatics.
Human evolutionary genetics studies how one human genome differs from another human genome, the evolutionary past that gave rise to the human genome, and its current effects. Differences between genomes have anthropological, medical, historical and forensic implications and applications. Genetic data can provide important insights into human evolution.

In molecular biology, SNORA2 is a non-coding RNA (ncRNA) which modifies other small nuclear RNAs (snRNAs). It is a member of the H/ACA class of small nucleolar RNA that guide the sites of modification of uridines to pseudouridines.
Minor allele frequency (MAF) is the frequency at which the second most common allele occurs in a given population. They play a surprising role in heritability since MAF variants which occur only once, known as "singletons", drive an enormous amount of selection.

The Single Nucleotide Polymorphism Database (dbSNP) is a free public archive for genetic variation within and across different species developed and hosted by the National Center for Biotechnology Information (NCBI) in collaboration with the National Human Genome Research Institute (NHGRI). Although the name of the database implies a collection of one class of polymorphisms only, it in fact contains a range of molecular variation: (1) SNPs, (2) short deletion and insertion polymorphisms (indels/DIPs), (3) microsatellite markers or short tandem repeats (STRs), (4) multinucleotide polymorphisms (MNPs), (5) heterozygous sequences, and (6) named variants. The dbSNP accepts apparently neutral polymorphisms, polymorphisms corresponding to known phenotypes, and regions of no variation. It was created in September 1998 to supplement GenBank, NCBI’s collection of publicly available nucleic acid and protein sequences.

HIKESHI is a protein important in lung and multicellular organismal development that, in humans, is encoded by the HIKESHI gene. HIKESHI is found on chromosome 11 in humans and chromosome 7 in mice. Similar sequences (orthologs) are found in most animal and fungal species. The mouse homolog, lethal gene on chromosome 7 Rinchik 6 protein is encoded by the l7Rn6 gene.
DECIPHER is a web-based resource and database of genomic variation data from analysis of patient DNA. It documents submicroscopic chromosome abnormalities and pathogenic sequence variants, from over 25000 patients and maps them to the human genome using Ensembl or UCSC Genome Browser. In addition it catalogues the clinical characteristics from each patient and maintains a database of microdeletion/duplication syndromes, together with links to relevant scientific reports and support groups.
Genomic structural variation is the variation in structure of an organism's chromosome, such as deletions, duplications, copy-number variants, insertions, inversions and translocations. Originally, a structure variation affects a sequence length about 1kb to 3Mb, which is larger than SNPs and smaller than chromosome abnormality. However, the operational range of structural variants has widened to include events > 50bp. Some structural variants are associated with genetic diseases, however most are not. Approximately 13% of the human genome is defined as structurally variant in the normal population, and there are at least 240 genes that exist as homozygous deletion polymorphisms in human populations, suggesting these genes are dispensable in humans. While humans carry a median of 3.6 Mbp in SNPs, a median of 8.9 Mbp is affected by structual variation which thus causes most genetic differences between humans in terms of raw sequence data.

A gene is said to be polymorphic if more than one allele occupies that gene's locus within a population. In addition to having more than one allele at a specific locus, each allele must also occur in the population at a rate of at least 1% to generally be considered polymorphic.
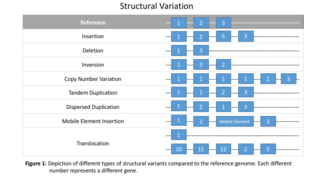
Structural variation in the human genome is operationally defined as genomic alterations, varying between individuals, that involve DNA segments larger than 1 kilo base (kb), and could be either microscopic or submicroscopic. This definition distinguishes them from smaller variants that are less than 1 kb in size such as short deletions, insertions, and single nucleotide variants.
ANNOVAR is a bioinformatics software tool for the interpretation and prioritization of single nucleotide variants (SNVs), insertions, deletions, and copy number variants (CNVs) of a given genome.