
In theoretical chemistry, a conjugated system is a system of connected p-orbitals with delocalized electrons in a molecule, which in general lowers the overall energy of the molecule and increases stability. It is conventionally represented as having alternating single and multiple bonds. Lone pairs, radicals or carbenium ions may be part of the system, which may be cyclic, acyclic, linear or mixed. The term "conjugated" was coined in 1899 by the German chemist Johannes Thiele.
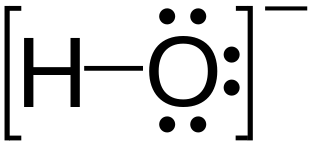
In chemistry, a lone pair refers to a pair of valence electrons that are not shared with another atom in a covalent bond and is sometimes called an unshared pair or non-bonding pair. Lone pairs are found in the outermost electron shell of atoms. They can be identified by using a Lewis structure. Electron pairs are therefore considered lone pairs if two electrons are paired but are not used in chemical bonding. Thus, the number of electrons in lone pairs plus the number of electrons in bonds equals the number of valence electrons around an atom.
In chemistry, π backbonding is a π-bonding interaction between a filled (or half filled) orbital of a transition metal atom and a vacant orbital on an adjacent ion or molecule. In this type of interaction, electrons from the metal are used to bond to the ligand, which dissipates excess negative charge and stabilizes the metal. It is common in transition metals with low oxidation states that have ligands such as carbon monoxide, olefins, or phosphines. The ligands involved in π backbonding can be broken into three groups: carbonyls and nitrogen analogs, alkenes and alkynes, and phosphines. Compounds where π backbonding is prominent include Ni(CO)4, Zeise's salt, and molybdenym and iron dinitrogen complexes.
In organic chemistry, a cycloaddition is a chemical reaction in which "two or more unsaturated molecules combine with the formation of a cyclic adduct in which there is a net reduction of the bond multiplicity". The resulting reaction is a cyclization reaction. Many but not all cycloadditions are concerted and thus pericyclic. Nonconcerted cycloadditions are not pericyclic. As a class of addition reaction, cycloadditions permit carbon–carbon bond formation without the use of a nucleophile or electrophile.
The 1,3-dipolar cycloaddition is a chemical reaction between a 1,3-dipole and a dipolarophile to form a five-membered ring. The earliest 1,3-dipolar cycloadditions were described in the late 19th century to the early 20th century, following the discovery of 1,3-dipoles. Mechanistic investigation and synthetic application were established in the 1960s, primarily through the work of Rolf Huisgen. Hence, the reaction is sometimes referred to as the Huisgen cycloaddition. 1,3-dipolar cycloaddition is an important route to the regio- and stereoselective synthesis of five-membered heterocycles and their ring-opened acyclic derivatives. The dipolarophile is typically an alkene or alkyne, but can be other pi systems. When the dipolarophile is an alkyne, aromatic rings are generally produced.

In organic chemistry, cheletropic reactions, also known as chelotropic reactions, are a type of pericyclic reaction. Specifically, cheletropic reactions are a subclass of cycloadditions. The key distinguishing feature of cheletropic reactions is that on one of the reagents, both new bonds are being made to the same atom.

In organic chemistry, hyperconjugation refers to the delocalization of electrons with the participation of bonds of primarily σ-character. Usually, hyperconjugation involves the interaction of the electrons in a sigma (σ) orbital with an adjacent unpopulated non-bonding p or antibonding σ* or π* orbitals to give a pair of extended molecular orbitals. However, sometimes, low-lying antibonding σ* orbitals may also interact with filled orbitals of lone pair character (n) in what is termed negative hyperconjugation. Increased electron delocalization associated with hyperconjugation increases the stability of the system. In particular, the new orbital with bonding character is stabilized, resulting in an overall stabilization of the molecule. Only electrons in bonds that are in the β position can have this sort of direct stabilizing effect — donating from a sigma bond on an atom to an orbital in another atom directly attached to it. However, extended versions of hyperconjugation can be important as well. The Baker–Nathan effect, sometimes used synonymously for hyperconjugation, is a specific application of it to certain chemical reactions or types of structures.
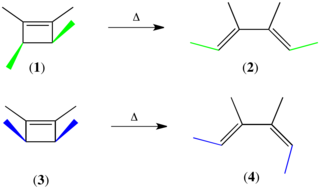
The Woodward–Hoffmann rules are a set of rules devised by Robert Burns Woodward and Roald Hoffmann to rationalize or predict certain aspects of the stereochemistry and activation energy of pericyclic reactions, an important class of reactions in organic chemistry. The rules originate in certain symmetries of the molecule's orbital structure that any molecular Hamiltonian conserves. Consequently, any symmetry-violating reaction must couple extensively to the environment; this imposes an energy barrier on its occurrence, and such reactions are called symmetry-forbidden. Their opposites are symmetry-allowed.

In theoretical chemistry, an antibonding orbital is a type of molecular orbital that weakens the chemical bond between two atoms and helps to raise the energy of the molecule relative to the separated atoms. Such an orbital has one or more nodes in the bonding region between the nuclei. The density of the electrons in the orbital is concentrated outside the bonding region and acts to pull one nucleus away from the other and tends to cause mutual repulsion between the two atoms. This is in contrast to a bonding molecular orbital, which has a lower energy than that of the separate atoms, and is responsible for chemical bonds.
Organogermanium chemistry is the science of chemical species containing one or more C–Ge bonds. Germanium shares group 14 in the periodic table with carbon, silicon, tin and lead. Historically, organogermanes are considered as nucleophiles and the reactivity of them is between that of organosilicon and organotin compounds. Some organogermanes have enhanced reactivity compared with their organosilicon and organoboron analogues in some cross-coupling reactions.

Disilyne is a silicon hydride with the formula Si
2H
2. Several isomers are possible, but none are sufficiently stable to be of practical value. Substituted disilynes contain a formal silicon–silicon triple bond and as such are sometimes written R2Si2 (where R is a substituent group). They are the silicon analogues of alkynes.

Germylenes are a class of germanium(II) compounds with the general formula :GeR2. They are heavier carbene analogs. However, unlike carbenes, whose ground state can be either singlet or triplet depending on the substituents, germylenes have exclusively a singlet ground state. Unprotected carbene analogs, including germylenes, has a dimerization nature. Free germylenes can be isolated under the stabilization of steric hindrance or electron donation. The synthesis of first stable free dialkyl germylene was reported by Jutzi, et al in 1991.
Boroles represent a class of molecules known as metalloles, which are heterocyclic 5-membered rings. As such, they can be viewed as structural analogs of cyclopentadiene, pyrrole or furan, with boron replacing a carbon, nitrogen and oxygen atom respectively. They are isoelectronic with the cyclopentadienyl cation C5H+5 or abbreviated as Cp+ and comprise four π electrons. Although Hückel's rule cannot be strictly applied to borole, it is considered to be antiaromatic due to having 4 π electrons. As a result, boroles exhibit unique electronic properties not found in other metalloles.
The nitrone-olefin (3+2) cycloaddition reaction is the combination of a nitrone with an alkene or alkyne to generate an isoxazoline or isoxazolidine via a (3+2) cycloaddition process. This reaction is a 1,3-dipolar cycloaddition, in which the nitrone acts as the 1,3-dipole, and the alkene or alkyne as the dipolarophile.
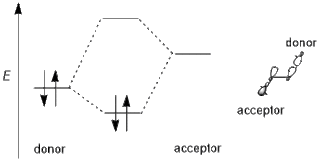
In chemistry, primarily organic and computational chemistry, a stereoelectronic effect is an effect on molecular geometry, reactivity, or physical properties due to spatial relationships in the molecules' electronic structure, in particular the interaction between atomic and/or molecular orbitals. Phrased differently, stereoelectronic effects can also be defined as the geometric constraints placed on the ground and/or transition states of molecules that arise from considerations of orbital overlap. Thus, a stereoelectronic effect explains a particular molecular property or reactivity by invoking stabilizing or destabilizing interactions that depend on the relative orientations of electrons in space.
Germanium(II) hydrides, also called germylene hydrides, are a class of Group 14 compounds consisting of low-valent germanium and a terminal hydride. They are also typically stabilized by an electron donor-acceptor interaction between the germanium atom and a large, bulky ligand.
In theoretical chemistry, the bonding orbital is used in molecular orbital (MO) theory to describe the attractive interactions between the atomic orbitals of two or more atoms in a molecule. In MO theory, electrons are portrayed to move in waves. When more than one of these waves come close together, the in-phase combination of these waves produces an interaction that leads to a species that is greatly stabilized. The result of the waves’ constructive interference causes the density of the electrons to be found within the binding region, creating a stable bond between the two species.

Trisilaallene is a subclass of silene derivatives where a central silicon atom forms double bonds with each of two terminal silicon atoms, with the generic formula R2Si=Si=SiR2. Trisilaallene is a silicon-based analog of an allene, but their chemical properties are markedly different.

Plumbylenes (or plumbylidenes) are divalent organolead(II) analogues of carbenes, with the general chemical formula, R2Pb, where R denotes a substituent. Plumbylenes possess 6 electrons in their valence shell, and are considered open shell species.
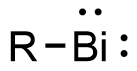
Bismuthinidenes are a class of organobismuth compounds, analogous to carbenes. These compounds have the general form R-Bi, with two lone pairs of electrons on the central bismuth(I) atom. Due to the unusually low valency and oxidation state of +1, most bismuthinidenes are reactive and unstable, though in recent decades, both transition metals and polydentate chelating Lewis base ligands have been employed to stabilize the low-valent bismuth(I) center through steric protection and π donation either in solution or in crystal structures. Lewis base-stabilized bismuthinidenes adopt a singlet ground state with an inert lone pair of electrons in the 6s orbital. A second lone pair in a 6p orbital and a single empty 6p orbital make Lewis base-stabilized bismuthinidenes ambiphilic.


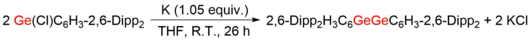
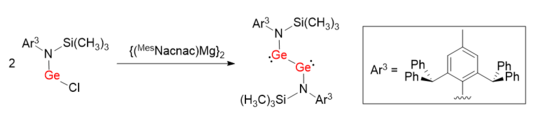

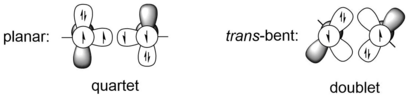



![[4+1] cycloaddition reaction of Ar GeGeAr with 1,3-dimethyl-1,3-butadiene. (4+1)cycloaddition of digermyne-pngversion.png](http://upload.wikimedia.org/wikipedia/commons/thumb/6/65/%284%2B1%29cycloaddition_of_digermyne-pngversion.png/460px-%284%2B1%29cycloaddition_of_digermyne-pngversion.png)

