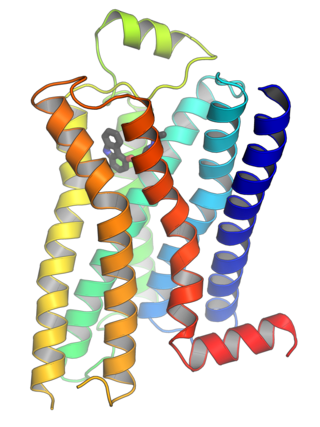
G protein-coupled receptors (GPCRs), also known as seven-(pass)-transmembrane domain receptors, 7TM receptors, heptahelical receptors, serpentine receptors, and G protein-linked receptors (GPLR), form a large group of evolutionarily related proteins that are cell surface receptors that detect molecules outside the cell and activate cellular responses. They are coupled with G proteins. They pass through the cell membrane seven times in the form of six loops of amino acid residues, which is why they are sometimes referred to as seven-transmembrane receptors. Ligands can bind either to the extracellular N-terminus and loops or to the binding site within transmembrane helices. They are all activated by agonists, although a spontaneous auto-activation of an empty receptor has also been observed.

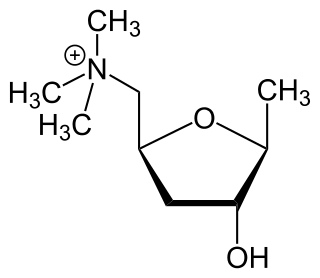
Acetylcholine (ACh) is an organic compound that functions in the brain and body of many types of animals as a neurotransmitter. Its name is derived from its chemical structure: it is an ester of acetic acid and choline. Parts in the body that use or are affected by acetylcholine are referred to as cholinergic.

An acetylcholine receptor or a cholinergic receptor is an integral membrane protein that responds to the binding of acetylcholine, a neurotransmitter.
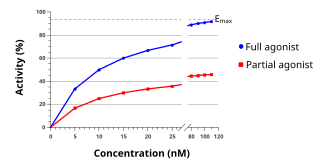
An agonist is a chemical that activates a receptor to produce a biological response. Receptors are cellular proteins whose activation causes the cell to modify what it is currently doing. In contrast, an antagonist blocks the action of the agonist, while an inverse agonist causes an action opposite to that of the agonist.

In biochemistry and pharmacology, receptors are chemical structures, composed of protein, that receive and transduce signals that may be integrated into biological systems. These signals are typically chemical messengers which bind to a receptor and produce physiological responses such as change in the electrical activity of a cell. For example, GABA, an inhibitory neurotransmitter, inhibits electrical activity of neurons by binding to GABAA receptors. There are three main ways the action of the receptor can be classified: relay of signal, amplification, or integration. Relaying sends the signal onward, amplification increases the effect of a single ligand, and integration allows the signal to be incorporated into another biochemical pathway.

A receptor antagonist is a type of receptor ligand or drug that blocks or dampens a biological response by binding to and blocking a receptor rather than activating it like an agonist. Antagonist drugs interfere in the natural operation of receptor proteins. They are sometimes called blockers; examples include alpha blockers, beta blockers, and calcium channel blockers. In pharmacology, antagonists have affinity but no efficacy for their cognate receptors, and binding will disrupt the interaction and inhibit the function of an agonist or inverse agonist at receptors. Antagonists mediate their effects by binding to the active site or to the allosteric site on a receptor, or they may interact at unique binding sites not normally involved in the biological regulation of the receptor's activity. Antagonist activity may be reversible or irreversible depending on the longevity of the antagonist–receptor complex, which, in turn, depends on the nature of antagonist–receptor binding. The majority of drug antagonists achieve their potency by competing with endogenous ligands or substrates at structurally defined binding sites on receptors.

A neuromuscular junction is a chemical synapse between a motor neuron and a muscle fiber.
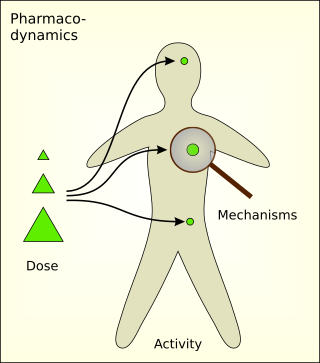
Pharmacodynamics (PD) is the study of the biochemical and physiologic effects of drugs. The effects can include those manifested within animals, microorganisms, or combinations of organisms.

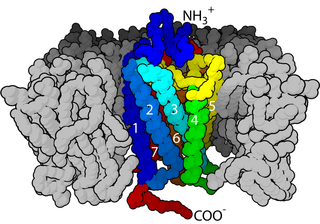
Nicotinic acetylcholine receptors, or nAChRs, are receptor polypeptides that respond to the neurotransmitter acetylcholine. Nicotinic receptors also respond to drugs such as the agonist nicotine. They are found in the central and peripheral nervous system, muscle, and many other tissues of many organisms. At the neuromuscular junction they are the primary receptor in muscle for motor nerve-muscle communication that controls muscle contraction. In the peripheral nervous system: (1) they transmit outgoing signals from the presynaptic to the postsynaptic cells within the sympathetic and parasympathetic nervous system, and (2) they are the receptors found on skeletal muscle that receive acetylcholine released to signal for muscular contraction. In the immune system, nAChRs regulate inflammatory processes and signal through distinct intracellular pathways. In insects, the cholinergic system is limited to the central nervous system.
Muscarinic acetylcholine receptors, or mAChRs, are acetylcholine receptors that form G protein-coupled receptor complexes in the cell membranes of certain neurons and other cells. They play several roles, including acting as the main end-receptor stimulated by acetylcholine released from postganglionic fibers. They are mainly found in the parasympathetic nervous system, but also have a role in the sympathetic nervous system in the control of sweat glands.
Neuropharmacology is the study of how drugs affect function in the nervous system, and the neural mechanisms through which they influence behavior. There are two main branches of neuropharmacology: behavioral and molecular. Behavioral neuropharmacology focuses on the study of how drugs affect human behavior (neuropsychopharmacology), including the study of how drug dependence and addiction affect the human brain. Molecular neuropharmacology involves the study of neurons and their neurochemical interactions, with the overall goal of developing drugs that have beneficial effects on neurological function. Both of these fields are closely connected, since both are concerned with the interactions of neurotransmitters, neuropeptides, neurohormones, neuromodulators, enzymes, second messengers, co-transporters, ion channels, and receptor proteins in the central and peripheral nervous systems. Studying these interactions, researchers are developing drugs to treat many different neurological disorders, including pain, neurodegenerative diseases such as Parkinson's disease and Alzheimer's disease, psychological disorders, addiction, and many others.

In biochemistry and pharmacology, a ligand is a substance that forms a complex with a biomolecule to serve a biological purpose. The etymology stems from Latin ligare, which means 'to bind'. In protein-ligand binding, the ligand is usually a molecule which produces a signal by binding to a site on a target protein. The binding typically results in a change of conformational isomerism (conformation) of the target protein. In DNA-ligand binding studies, the ligand can be a small molecule, ion, or protein which binds to the DNA double helix. The relationship between ligand and binding partner is a function of charge, hydrophobicity, and molecular structure.

Neuromuscular-blocking drugs, or Neuromuscular blocking agents (NMBAs), block transmission at the neuromuscular junction, causing paralysis of the affected skeletal muscles. This is accomplished via their action on the post-synaptic acetylcholine (Nm) receptors.
The Cys-loop ligand-gated ion channel superfamily is composed of nicotinic acetylcholine, GABAA, GABAA-ρ, glycine, 5-HT3, and zinc-activated (ZAC) receptors. These receptors are composed of five protein subunits which form a pentameric arrangement around a central pore. There are usually 2 alpha subunits and 3 other beta, gamma, or delta subunits (some consist of 5 alpha subunits). The name of the family refers to a characteristic loop formed by 13 highly conserved amino acids between two cysteine (Cys) residues, which form a disulfide bond near the N-terminal extracellular domain.
A nicotinic agonist is a drug that mimics the action of acetylcholine (ACh) at nicotinic acetylcholine receptors (nAChRs). The nAChR is named for its affinity for nicotine.
Receptor theory is the application of receptor models to explain drug behavior. Pharmacological receptor models preceded accurate knowledge of receptors by many years. John Newport Langley and Paul Ehrlich introduced the concept that receptors can mediate drug action at the beginning of the 20th century. Alfred Joseph Clark was the first to quantify drug-induced biological responses. So far, nearly all of the quantitative theoretical modelling of receptor function has centred on ligand-gated ion channels and G protein-coupled receptors.
Cell surface receptors are receptors that are embedded in the plasma membrane of cells. They act in cell signaling by receiving extracellular molecules. They are specialized integral membrane proteins that allow communication between the cell and the extracellular space. The extracellular molecules may be hormones, neurotransmitters, cytokines, growth factors, cell adhesion molecules, or nutrients; they react with the receptor to induce changes in the metabolism and activity of a cell. In the process of signal transduction, ligand binding affects a cascading chemical change through the cell membrane.
In pharmacology and biochemistry, allosteric modulators are a group of substances that bind to a receptor to change that receptor's response to stimuli. Some of them, like benzodiazepines or alcohol, function as psychoactive drugs. The site that an allosteric modulator binds to is not the same one to which an endogenous agonist of the receptor would bind. Modulators and agonists can both be called receptor ligands.
A receptor modulator, or receptor ligand, is a general term for a substance, endogenous or exogenous, that binds to and regulates the activity of chemical receptors. They are ligands that can act on different parts of receptors and regulate activity in a positive, negative, or neutral direction with varying degrees of efficacy. Categories of these modulators include receptor agonists and receptor antagonists, as well as receptor partial agonists, inverse agonists, orthosteric modulators, and allosteric modulators, Examples of receptor modulators in modern medicine include CFTR modulators, selective androgen receptor modulators (SARMs), and muscarinic ACh receptor modulators.
Autonomic drugs are substances that can either inhibit or enhance the functions of the parasympathetic and sympathetic nervous systems. This type of drug can be used to treat a wide range of diseases an disorders, including glaucoma, asthma, and disorders of the urinary, gastrointestinal and circulatory systems.
