
Alternative splicing, or alternative RNA splicing, or differential splicing, is an alternative splicing process during gene expression that allows a single gene to code for multiple proteins. In this process, particular exons of a gene may be included within or excluded from the final, processed messenger RNA (mRNA) produced from that gene. This means the exons are joined in different combinations, leading to different (alternative) mRNA strands. Consequently, the proteins translated from alternatively spliced mRNAs usually contain differences in their amino acid sequence and, often, in their biological functions.
In computational biology, gene prediction or gene finding refers to the process of identifying the regions of genomic DNA that encode genes. This includes protein-coding genes as well as RNA genes, but may also include prediction of other functional elements such as regulatory regions. Gene finding is one of the first and most important steps in understanding the genome of a species once it has been sequenced.
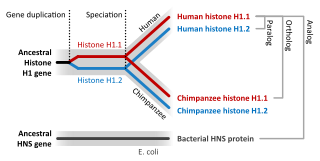
Sequence homology is the biological homology between DNA, RNA, or protein sequences, defined in terms of shared ancestry in the evolutionary history of life. Two segments of DNA can have shared ancestry because of three phenomena: either a speciation event (orthologs), or a duplication event (paralogs), or else a horizontal gene transfer event (xenologs).

UniProt is a freely accessible database of protein sequence and functional information, many entries being derived from genome sequencing projects. It contains a large amount of information about the biological function of proteins derived from the research literature. It is maintained by the UniProt consortium, which consists of several European bioinformatics organisations and a foundation from Washington, DC, United States.
The European Bioinformatics Institute (EMBL-EBI) is an intergovernmental organization (IGO) which, as part of the European Molecular Biology Laboratory (EMBL) family, focuses on research and services in bioinformatics. It is located on the Wellcome Genome Campus in Hinxton near Cambridge, and employs over 600 full-time equivalent (FTE) staff. Institute leaders such as Rolf Apweiler, Alex Bateman, Ewan Birney, and Guy Cochrane, an adviser on the National Genomics Data Center Scientific Advisory Board, serve as part of the international research network of the BIG Data Center at the Beijing Institute of Genomics.
The completion of the human genome sequencing in the early 2000s was a turning point in genomics research. Scientists have conducted series of research into the activities of genes and the genome as a whole. The human genome contains around 3 billion base pairs nucleotide, and the huge quantity of data created necessitates the development of an accessible tool to explore and interpret this information in order to investigate the genetic basis of disease, evolution, and biological processes. The field of genomics has continued to grow, with new sequencing technologies and computational tool making it easier to study the genome.
Protein function prediction methods are techniques that bioinformatics researchers use to assign biological or biochemical roles to proteins. These proteins are usually ones that are poorly studied or predicted based on genomic sequence data. These predictions are often driven by data-intensive computational procedures. Information may come from nucleic acid sequence homology, gene expression profiles, protein domain structures, text mining of publications, phylogenetic profiles, phenotypic profiles, and protein-protein interaction. Protein function is a broad term: the roles of proteins range from catalysis of biochemical reactions to transport to signal transduction, and a single protein may play a role in multiple processes or cellular pathways.

In molecular biology and genetics, DNA annotation or genome annotation is the process of describing the structure and function of the components of a genome, by analyzing and interpreting them in order to extract their biological significance and understand the biological processes in which they participate. Among other things, it identifies the locations of genes and all the coding regions in a genome and determines what those genes do.
ASPicDB is a database of human protein variants generated by alternative splicing, a process by which the exons of the RNA produced by transcription of a gene are reconnected in multiple ways during RNA splicing.
Alternative Splicing Annotation Project (ASAP) in computational biology was a database for alternative splicing data maintained by the University of California from 2003 to 2013. The purpose of ASAP was to provide a source for data mining projects by consolidating the information generated by genomics and proteomics researchers.
The Alternative Splicing and Transcript Diversity database (ASTD) was a database of transcript variants maintained by the European Bioinformatics Institute from 2008 to 2012. It contained transcription initiation, polyadenylation and splicing variant data.
ChimerDB in computational biology is a database of fusion sequences.
The Consensus Coding Sequence (CCDS) Project is a collaborative effort to maintain a dataset of protein-coding regions that are identically annotated on the human and mouse reference genome assemblies. The CCDS project tracks identical protein annotations on the reference mouse and human genomes with a stable identifier, and ensures that they are consistently represented by the National Center for Biotechnology Information (NCBI), Ensembl, and UCSC Genome Browser. The integrity of the CCDS dataset is maintained through stringent quality assurance testing and on-going manual curation.
The Intronerator is a database of alternatively spliced genes and a database of introns for Caenorhabditis elegans.
SpliceInfo is a database for the four major alternative-splicing modes in the human genome.
De novo transcriptome assembly is the de novo sequence assembly method of creating a transcriptome without the aid of a reference genome.
Chimeric RNA, sometimes referred to as a fusion transcript, is composed of exons from two or more different genes that have the potential to encode novel proteins. These mRNAs are different from those produced by conventional splicing as they are produced by two or more gene loci.
WormBase is an online biological database about the biology and genome of the nematode model organism Caenorhabditis elegans and contains information about other related nematodes. WormBase is used by the C. elegans research community both as an information resource and as a place to publish and distribute their results. The database is regularly updated with new versions being released every two months. WormBase is one of the organizations participating in the Generic Model Organism Database (GMOD) project.
Single nucleotide polymorphism annotation is the process of predicting the effect or function of an individual SNP using SNP annotation tools. In SNP annotation the biological information is extracted, collected and displayed in a clear form amenable to query. SNP functional annotation is typically performed based on the available information on nucleic acid and protein sequences.