
Catalysis is the process of change in rate of a chemical reaction by adding a substance known as a catalyst. Catalysts are not consumed by the reaction and remain unchanged after it. If the reaction is rapid and the catalyst recycles quickly, very small amounts of catalyst often suffice; mixing, surface area, and temperature are important factors in reaction rate. Catalysts generally react with one or more reactants to form intermediates that subsequently give the final reaction product, in the process of regenerating the catalyst.

A chemical bond is a lasting attraction between atoms or ions that enables the formation of molecules, crystals, and other structures. The bond may result from the electrostatic force between oppositely charged ions as in ionic bonds, or through the sharing of electrons as in covalent bonds. The strength of chemical bonds varies considerably; there are "strong bonds" or "primary bonds" such as covalent, ionic and metallic bonds, and "weak bonds" or "secondary bonds" such as dipole–dipole interactions, the London dispersion force, and hydrogen bonding.
Computational chemistry is a branch of chemistry that uses computer simulation to assist in solving chemical problems. It uses methods of theoretical chemistry, incorporated into computer programs, to calculate the structures and properties of molecules, groups of molecules, and solids. It is essential because, apart from relatively recent results concerning the hydrogen molecular ion, the quantum many-body problem cannot be solved analytically, much less in closed form. While computational results normally complement the information obtained by chemical experiments, it can in some cases predict hitherto unobserved chemical phenomena. It is widely used in the design of new drugs and materials.

A chemical reaction is a process that leads to the chemical transformation of one set of chemical substances to another. Classically, chemical reactions encompass changes that only involve the positions of electrons in the forming and breaking of chemical bonds between atoms, with no change to the nuclei, and can often be described by a chemical equation. Nuclear chemistry is a sub-discipline of chemistry that involves the chemical reactions of unstable and radioactive elements where both electronic and nuclear changes can occur.
Quantum chemistry, also called molecular quantum mechanics, is a branch of physical chemistry focused on the application of quantum mechanics to chemical systems, particularly towards the quantum-mechanical calculation of electronic contributions to physical and chemical properties of molecules, materials, and solutions at the atomic level. These calculations include systematically applied approximations intended to make calculations computationally feasible while still capturing as much information about important contributions to the computed wave functions as well as to observable properties such as structures, spectra, and thermodynamic properties. Quantum chemistry is also concerned with the computation of quantum effects on molecular dynamics and chemical kinetics.

In chemistry and physics, activation energy is the minimum amount of energy that must be provided for compounds to result in a chemical reaction. The activation energy (Ea) of a reaction is measured in joules per mole (J/mol), kilojoules per mole (kJ/mol) or kilocalories per mole (kcal/mol). Activation energy can be thought of as the magnitude of the potential barrier (sometimes called the energy barrier) separating minima of the potential energy surface pertaining to the initial and final thermodynamic state. For a chemical reaction to proceed at a reasonable rate, the temperature of the system should be high enough such that there exists an appreciable number of molecules with translational energy equal to or greater than the activation energy. The term "activation energy" was introduced in 1889 by the Swedish scientist Svante Arrhenius.

Molecular dynamics (MD) is a computer simulation method for analyzing the physical movements of atoms and molecules. The atoms and molecules are allowed to interact for a fixed period of time, giving a view of the dynamic "evolution" of the system. In the most common version, the trajectories of atoms and molecules are determined by numerically solving Newton's equations of motion for a system of interacting particles, where forces between the particles and their potential energies are often calculated using interatomic potentials or molecular mechanical force fields. The method is applied mostly in chemical physics, materials science, and biophysics.
Chemical kinetics, also known as reaction kinetics, is the branch of physical chemistry that is concerned with understanding the rates of chemical reactions. It is different from chemical thermodynamics, which deals with the direction in which a reaction occurs but in itself tells nothing about its rate. Chemical kinetics includes investigations of how experimental conditions influence the speed of a chemical reaction and yield information about the reaction's mechanism and transition states, as well as the construction of mathematical models that also can describe the characteristics of a chemical reaction.
In chemistry, reactivity is the impulse for which a chemical substance undergoes a chemical reaction, either by itself or with other materials, with an overall release of energy.
In physical organic chemistry, a kinetic isotope effect (KIE) is the change in the reaction rate of a chemical reaction when one of the atoms in the reactants is replaced by one of its isotopes. Formally, it is the ratio of rate constants for the reactions involving the light (kL) and the heavy (kH) isotopically substituted reactants (isotopologues):

A potential energy surface (PES) describes the energy of a system, especially a collection of atoms, in terms of certain parameters, normally the positions of the atoms. The surface might define the energy as a function of one or more coordinates; if there is only one coordinate, the surface is called a potential energy curve or energy profile. An example is the Morse/Long-range potential.

In the context of chemistry and molecular modelling, a force field is a computational method that is used to estimate the forces between atoms within molecules and also between molecules. More precisely, the force field refers to the functional form and parameter sets used to calculate the potential energy of a system of atoms or coarse-grained particles in molecular mechanics, molecular dynamics, or Monte Carlo simulations. The parameters for a chosen energy function may be derived from experiments in physics and chemistry, calculations in quantum mechanics, or both. Force fields are interatomic potentials and utilize the same concept as force fields in classical physics, with the difference that the force field parameters in chemistry describe the energy landscape, from which the acting forces on every particle are derived as a gradient of the potential energy with respect to the particle coordinates.
The bond valencemethod or mean method is a popular method in coordination chemistry to estimate the oxidation states of atoms. It is derived from the bond valence model, which is a simple yet robust model for validating chemical structures with localized bonds or used to predict some of their properties. This model is a development of Pauling's rules.
The 18-electron rule is a chemical rule of thumb used primarily for predicting and rationalizing formulas for stable transition metal complexes, especially organometallic compounds. The rule is based on the fact that the valence orbitals in the electron configuration of transition metals consist of five (n−1)d orbitals, one ns orbital, and three np orbitals, where n is the principal quantum number. These orbitals can collectively accommodate 18 electrons as either bonding or non-bonding electron pairs. This means that the combination of these nine atomic orbitals with ligand orbitals creates nine molecular orbitals that are either metal-ligand bonding or non-bonding. When a metal complex has 18 valence electrons, it is said to have achieved the same electron configuration as the noble gas in the period, lending stability to the complex. Transition metal complexes that deviate from the rule are often interesting or useful because they tend to be more reactive. The rule is not helpful for complexes of metals that are not transition metals. The rule was first proposed by American chemist Irving Langmuir in 1921.
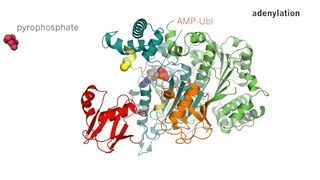
Enzyme catalysis is the increase in the rate of a process by a biological molecule, an "enzyme". Most enzymes are proteins, and most such processes are chemical reactions. Within the enzyme, generally catalysis occurs at a localized site, called the active site.

In chemistry, transition state theory (TST) explains the reaction rates of elementary chemical reactions. The theory assumes a special type of chemical equilibrium (quasi-equilibrium) between reactants and activated transition state complexes.

Arieh Warshel is an Israeli-American biochemist and biophysicist. He is a pioneer in computational studies on functional properties of biological molecules, Distinguished Professor of Chemistry and Biochemistry, and holds the Dana and David Dornsife Chair in Chemistry at the University of Southern California. He received the 2013 Nobel Prize in Chemistry, together with Michael Levitt and Martin Karplus for "the development of multiscale models for complex chemical systems".
This glossary of chemistry terms is a list of terms and definitions relevant to chemistry, including chemical laws, diagrams and formulae, laboratory tools, glassware, and equipment. Chemistry is a physical science concerned with the composition, structure, and properties of matter, as well as the changes it undergoes during chemical reactions; it features an extensive vocabulary and a significant amount of jargon.

In theoretical chemistry, an energy profile is a theoretical representation of a chemical reaction or process as a single energetic pathway as the reactants are transformed into products. This pathway runs along the reaction coordinate, which is a parametric curve that follows the pathway of the reaction and indicates its progress; thus, energy profiles are also called reaction coordinate diagrams. They are derived from the corresponding potential energy surface (PES), which is used in computational chemistry to model chemical reactions by relating the energy of a molecule(s) to its structure.
Q is a computer software package for molecular dynamics (MD) simulation. Unlike other MD codes, it has specialized since its conception on three specific types of free energy calculations. These calculations are based on the methods: empirical valence bond (EVB), free energy perturbation (FEP), and linear interaction energy (LIE), as well as, more recently, also path integral calculations using the bisection quantum classical path (BQCP) approach.

