
Holoprosencephaly (HPE) is a cephalic disorder in which the prosencephalon fails to develop into two hemispheres, typically occurring between the 18th and 28th day of gestation. Normally, the forebrain is formed and the face begins to develop in the fifth and sixth weeks of human pregnancy. The condition also occurs in other species.

Cebocephaly is a developmental anomaly that is part of a group of defects called holoprosencephaly. Cebocephaly involves the presence of two separate eyes set close together and a small, flat nose with a single nostril. It may be diagnosed before or after birth. It has a very poor prognosis, with most affected infants dying soon after birth. It is very rare, having been estimated to affect around 1 in 40,000 deliveries.
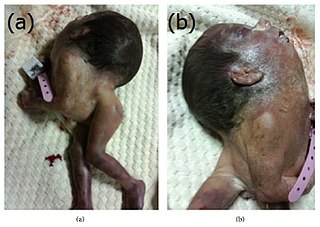
Iniencephaly is a rare type of cephalic disorder characterised by three common characteristics: a defect to the occipital bone, spina bifida of the cervical vertebrae and retroflexion of the head on the cervical spine. Stillbirth is the most common outcome, with a few rare examples of live birth, after which death invariably occurs within a short time.
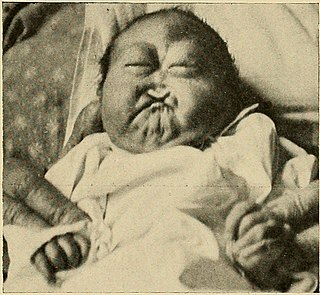
Patau syndrome is a syndrome caused by a chromosomal abnormality, in which some or all of the cells of the body contain extra genetic material from chromosome 13. The extra genetic material disrupts normal development, causing multiple and complex organ defects.

Trisomy 18, also known as Edwards syndrome, is a genetic disorder caused by the presence of a third copy of all or part of chromosome 18. Many parts of the body are affected. Babies are often born small and have heart defects. Other features include a small head, small jaw, clenched fists with overlapping fingers, and severe intellectual disability.
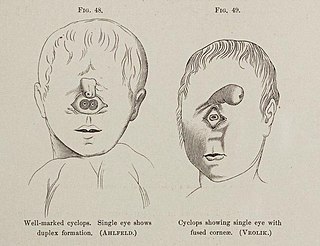
Cyclopia, also known as alobar holoprosencephaly, is the most extreme form of holoprosencephaly and is a congenital disorder characterized by the failure of the embryonic prosencephalon to properly divide the orbits of the eye into two cavities. Its incidence is 1 in 16,000 in born animals and 1 in 200 in miscarried fetuses.

Cri du chat syndrome is a rare genetic disorder due to a partial chromosome deletion on chromosome 5. Its name is a French term referring to the characteristic cat-like cry of affected children. It was first described by Jérôme Lejeune in 1963. The condition affects an estimated 1 in 50,000 live births across all ethnicities and is more common in females by a 4:3 ratio.

Otocephaly, also known as agnathia–otocephaly complex, is a very rare and lethal cephalic disorder characterized by the absence of the mandible (agnathia), with the ears fused together just below the chin (synotia). It is caused by a disruption to the development of the first branchial arch. It occurs in every 1 in 70,000 embryos.
The Pallister–Killian syndrome (PKS), also termed tetrasomy 12p mosaicism or the Pallister mosaic aneuploidy syndrome, is an extremely rare and severe genetic disorder. PKS is due to the presence of an extra and abnormal chromosome termed a small supernumerary marker chromosome (sSMC). sSMCs contain copies of genetic material from parts of virtually any other chromosome and, depending on the genetic material they carry, can cause various genetic disorders and neoplasms. The sSMC in PKS consists of multiple copies of the short arm of chromosome 12. Consequently, the multiple copies of the genetic material in the sSMC plus the two copies of this genetic material in the two normal chromosome 12's are overexpressed and thereby cause the syndrome. Due to a form of genetic mosaicism, however, individuals with PKS differ in the tissue distributions of their sSMC and therefore show different syndrome-related birth defects and disease severities. For example, individuals with the sSMC in their heart tissue are likely to have cardiac structural abnormalities while those without this sSMC localization have a structurally normal heart.

Triploid syndrome, also called triploidy, is a chromosomal disorder in which a fetus has three copies of every chromosome instead of the normal two. If this occurs in only some cells, it is called mosaic triploidy and is less severe.

Trisomy 22 is a chromosomal disorder in which three copies of chromosome 22 are present rather than two. It is a frequent cause of spontaneous abortion during the first trimester of pregnancy. Progression to the second trimester and live birth are rare. This disorder is found in individuals with an extra copy or a variation of chromosome 22 in some or all cells of their bodies.

Frontonasal dysplasia (FND) is a congenital malformation of the midface. For the diagnosis of FND, a patient should present at least two of the following characteristics: hypertelorism, a wide nasal root, vertical midline cleft of the nose and/or upper lip, cleft of the wings of the nose, malformed nasal tip, encephalocele or V-shaped hair pattern on the forehead. The cause of FND remains unknown. FND seems to be sporadic (random) and multiple environmental factors are suggested as possible causes for the syndrome. However, in some families multiple cases of FND were reported, which suggests a genetic cause of FND.

Roberts syndrome, or sometimes called pseudothalidomide syndrome, is an extremely rare autosomal recessive genetic disorder that is characterized by mild to severe prenatal retardation or disruption of cell division, leading to malformation of the bones in the skull, face, arms, and legs.
A malformative syndrome is a recognizable pattern of congenital anomalies that are known or thought to be causally related.
A facial cleft is an opening or gap in the face, or a malformation of a part of the face. Facial clefts is a collective term for all sorts of clefts. All structures like bone, soft tissue, skin etc. can be affected. Facial clefts are extremely rare congenital anomalies. There are many variations of a type of clefting and classifications are needed to describe and classify all types of clefting. Facial clefts hardly ever occur isolated; most of the time there is an overlap of adjacent facial clefts.
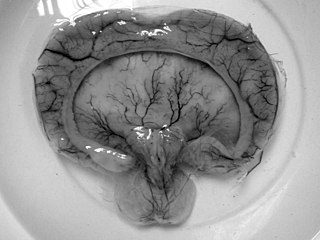
Young–Madders syndrome, alternatively known as Pseudotrisomy 13 syndrome or holoprosencephaly–polydactyly syndrome, is a genetic disorder resulting from defective and duplicated chromosomes which result in holoprosencephaly, polydactyly, facial malformations and intellectual disability, with a significant variance in the severity of symptoms being seen across known cases. Many cases often suffer with several other genetic disorders, and some have presented with hypoplasia, cleft lip, cardiac lesions and other heart defects. In one case in 1991 and another in 2000 the condition was found in siblings who were the product of incest. Many cases are diagnosed prenatally and often in siblings. Cases are almost fatal in the prenatal stage with babies being stillborn.

13q deletion syndrome is a rare genetic disease caused by the deletion of some or all of the large arm of human chromosome 13. Depending upon the size and location of the deletion on chromosome 13, the physical and mental manifestations will vary. It has the potential to cause intellectual disability and congenital malformations that affect a variety of organ systems. Because of the rarity of the disease in addition to the variations in the disease, the specific genes that cause this disease are unknown. This disease is also known as:
XK aprosencephaly is an extremely rare congenital disorder characterized by the absence of the embryonic forebrain. Because the prosencephalon gives way to the cerebral cortex, survival with aprosencephaly is not possible outside utero. The external symptoms are similar to holoprosencephaly, a related disorder, including a smaller than normal head (microcephaly), small eyeballs (microphthalmia), a small mouth (microstomia), anal atresia, and abnormalities of the external genitalia, radius, nostrils, and pharynx (throat).

Pai syndrome, also known as Median cleft of the upper lip-corpus callosum lipoma-midline facial cutaneous polyps syndrome, is a very rare genetic disorder which is characterized by nervous system, cutaneous, ocular, nasal and bucal anomalies with facial dysmorphisms.
