
Creutzfeldt–Jakob disease (CJD), also known as subacute spongiform encephalopathy or neurocognitive disorder due to prion disease, is a fatal neurodegenerative disease. Early symptoms include memory problems, behavioral changes, poor coordination, and visual disturbances. Later symptoms include dementia, involuntary movements, blindness, weakness, and coma. About 70% of people die within a year of diagnosis. The name "Creutzfeldt–Jakob disease" was introduced by Walther Spielmeyer in 1922, after the German neurologists Hans Gerhard Creutzfeldt and Alfons Maria Jakob.

Dementia is a syndrome associated with many neurodegenerative diseases, characterized by a general decline in cognitive abilities that affects a person's ability to perform everyday activities. This typically involves problems with memory, thinking, behavior, and motor control. Aside from memory impairment and a disruption in thought patterns, the most common symptoms of dementia include emotional problems, difficulties with language, and decreased motivation. The symptoms may be described as occurring in a continuum over several stages. Dementia ultimately has a significant effect on the individual, their caregivers, and their social relationships in general. A diagnosis of dementia requires the observation of a change from a person's usual mental functioning and a greater cognitive decline than might be caused by the normal aging process.

Fatal insomnia is an extremely rare neurodegenerative prion disease that results in trouble sleeping as its hallmark symptom. The majority of cases are familial, stemming from a mutation in the PRNP gene, with the remainder of cases occurring sporadically. The problems with sleeping typically start out gradually and worsen over time. Eventually, the patient will succumb to total insomnia, most often leading to other symptoms such as speech problems, coordination problems, and dementia. It results in death within a few months to a few years, and there is no known disease-modifying treatment.
Degenerative disease is the result of a continuous process based on degenerative cell changes, affecting tissues or organs, which will increasingly deteriorate over time.

The tau proteins form a group of six highly soluble protein isoforms produced by alternative splicing from the gene MAPT. They have roles primarily in maintaining the stability of microtubules in axons and are abundant in the neurons of the central nervous system (CNS), where the cerebral cortex has the highest abundance. They are less common elsewhere but are also expressed at very low levels in CNS astrocytes and oligodendrocytes.

Gerstmann–Sträussler–Scheinker syndrome (GSS) is an extremely rare, always fatal neurodegenerative disease that affects patients from 20 to 60 years in age. It is exclusively heritable, and is found in only a few families all over the world. It is, however, classified with the transmissible spongiform encephalopathies (TSE) due to the causative role played by PRNP, the human prion protein. GSS was first reported by the Austrian physicians Josef Gerstmann, Ernst Sträussler and Ilya Scheinker in 1936.
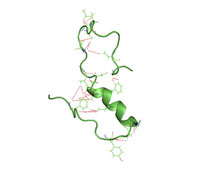
Amyloid beta denotes peptides of 36–43 amino acids that are the main component of the amyloid plaques found in the brains of people with Alzheimer's disease. The peptides derive from the amyloid-beta precursor protein (APP), which is cleaved by beta secretase and gamma secretase to yield Aβ in a cholesterol-dependent process and substrate presentation. Both neurons and oligodendrocytes produce and release Aβ in the brain, contributing to formation of amyloid plaques. Aβ molecules can aggregate to form flexible soluble oligomers which may exist in several forms. It is now believed that certain misfolded oligomers can induce other Aβ molecules to also take the misfolded oligomeric form, leading to a chain reaction akin to a prion infection. The oligomers are toxic to nerve cells. The other protein implicated in Alzheimer's disease, tau protein, also forms such prion-like misfolded oligomers, and there is some evidence that misfolded Aβ can induce tau to misfold.

Amyloid plaques are extracellular deposits of amyloid beta (Aβ) protein that present mainly in the grey matter of the brain. Degenerative neuronal elements and an abundance of microglia and astrocytes can be associated with amyloid plaques. Some plaques occur in the brain as a result of aging, but large numbers of plaques and neurofibrillary tangles are characteristic features of Alzheimer's disease. The plaques are highly variable in shape and size; in tissue sections immunostained for Aβ, they comprise a log-normal size distribution curve, with an average plaque area of 400-450 square micrometers (μm2). The smallest plaques, which often consist of diffuse deposits of Aβ, are particularly numerous. Plaques form when Aβ misfolds and aggregates into oligomers and longer polymers, the latter of which are characteristic of amyloid.

The major prion protein (PrP) is encoded in the human body by the PRNP gene also known as CD230. Expression of the protein is most predominant in the nervous system but occurs in many other tissues throughout the body.
A neurodegenerative disease is caused by the progressive loss of neurons, in the process known as neurodegeneration. Neuronal damage may also ultimately result in their death. Neurodegenerative diseases include amyotrophic lateral sclerosis, multiple sclerosis, Parkinson's disease, Alzheimer's disease, Huntington's disease, multiple system atrophy, tauopathies, and prion diseases. Neurodegeneration can be found in the brain at many different levels of neuronal circuitry, ranging from molecular to systemic. Because there is no known way to reverse the progressive degeneration of neurons, these diseases are considered to be incurable; however research has shown that the two major contributing factors to neurodegeneration are oxidative stress and inflammation. Biomedical research has revealed many similarities between these diseases at the subcellular level, including atypical protein assemblies and induced cell death. These similarities suggest that therapeutic advances against one neurodegenerative disease might ameliorate other diseases as well.

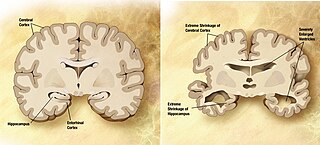
Alzheimer's disease (AD) is a neurodegenerative disease that usually starts slowly and progressively worsens. It is the cause of 60–70% of cases of dementia. The most common early symptom is difficulty in remembering recent events. As the disease advances, symptoms can include problems with language, disorientation, mood swings, loss of motivation, self-neglect, and behavioral issues. As a person's condition declines, they often withdraw from family and society. Gradually, bodily functions are lost, ultimately leading to death. Although the speed of progression can vary, the average life expectancy following diagnosis is three to twelve years.
Early-onset Alzheimer's disease (EOAD), also called younger-onset Alzheimer's disease (YOAD), is Alzheimer's disease diagnosed before the age of 65. It is an uncommon form of Alzheimer's, accounting for only 5–10% of all Alzheimer's cases. About 60% have a positive family history of Alzheimer's and 13% of them are inherited in an autosomal dominant manner. Most cases of early-onset Alzheimer's share the same traits as the "late-onset" form and are not caused by known genetic mutations. Little is understood about how it starts.
The National Prion Clinic (UK) is part of the University College London Hospitals NHS Foundation Trust. Its aim is to diagnose and treat patients with any form of human prion disease (Creutzfeldt-Jakob disease, CJD). In addition, the clinic facilitates research in diagnostics and therapeutics, organises clinical trials, and counsels those with an increased genetic risk of the disease.

Variably protease-sensitive prionopathy (VPSPr) is a sporadic prion protein disease first described in an abstract for a conference on prions in 2006, and this study was published in a 2008 report on 11 cases. The study was conducted by Gambetti P., Zou W.Q., and coworkers from the United States National Prion Disease Pathology Surveillance Center. It was first identified as a distinct disease in 2010 by Zou W.Q. and coworkers from the United States National Prion Disease Pathology Surveillance Center.

Rosalind Ridley is a British psychologist and researcher who was head of the Medical Research Council Comparative Cognition Research Team in the Department of Psychology, Cambridge, UK, until 2005. She was a fellow of Newnham College, Cambridge from 1995–2010 and Vice-Principal from 2000–2005. She holds the privileges of a Fellow Emerita at Newnham College.
Huntington's disease-like syndromes are a family of inherited neurodegenerative diseases that closely resemble Huntington's disease (HD) in that they typically produce a combination of chorea, cognitive decline or dementia and behavioural or psychiatric problems.

Giovanna Rachele Mallucci is van Geest Professor of Clinical Neurosciences at the University of Cambridge in England and associate director of the UK Dementia Research Institute at the University of Cambridge. She is a specialist in neurodegenerative diseases.
Michael D. Geschwind is a professor of neurology at the UCSF Memory and Aging Center (MAC), specializing in neurodegenerative disorders.
Familial Danish dementia is an extremely rare, neurodegenerative disease characterized by progressive cataracts, loss of hearing, cerebellar ataxia, paranoid psychosis, and dementia. Neuropathological hallmarks include extensive atrophy of all areas of the brain, chronic diffuse encephalopathy, and the presence of exceedingly thin and nearly totally demyelinated cranial nerves.
Gerard David Schellenberg is an academic neuropathologist who specializes in the research of Alzheimer's disease. He is the director of Penn Neurodegeneration Genomics Center as well as a professor of Pathology and Laboratory Medicine at the University of Pennsylvania. He is a leading contributor to Alzheimer's disease research.