Related Research Articles

Brugada syndrome (BrS) is a genetic disorder in which the electrical activity of the heart is abnormal due to channelopathy. It increases the risk of abnormal heart rhythms and sudden cardiac death. Those affected may have episodes of syncope. The abnormal heart rhythms seen in those with Brugada syndrome often occur at rest. They may be triggered by a fever.

Sodium channel protein type 5 subunit alpha, also known as NaV1.5 is an integral membrane protein and tetrodotoxin-resistant voltage-gated sodium channel subunit. NaV1.5 is found primarily in cardiac muscle, where it mediates the fast influx of Na+-ions (INa) across the cell membrane, resulting in the fast depolarization phase of the cardiac action potential. As such, it plays a major role in impulse propagation through the heart. A vast number of cardiac diseases is associated with mutations in NaV1.5 (see paragraph genetics). SCN5A is the gene that encodes the cardiac sodium channel NaV1.5.

The glycerol-3-phosphate shuttle is a mechanism that regenerates NAD+ from NADH, a by-product of glycolysis. The shuttle consists of the sequential activity of two proteins: GPD1 which transfers an electron pair from NADH to dihydroxyacetone phosphate (DHAP), forming glycerol-3-phosphate (G3P) and regenerating NAD+ needed to generate energy via glycolysis. The mitochondrial inner membrane protein GPD2 catalyzes the oxidation of G3P, regenerating DHAP in the cytosol and forming FADH2 in the mitochondrial matrix. In mammals, its activity in transporting reducing equivalents across the mitochondrial membrane is considered secondary to the malate-aspartate shuttle.

Trifunctional enzyme subunit alpha, mitochondrial also known as hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase/enoyl-CoA hydratase, alpha subunit is a protein that in humans is encoded by the HADHA gene. Mutations in HADHA have been associated with trifunctional protein deficiency or long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency.

Trifunctional enzyme subunit beta, mitochondrial (TP-beta) also known as 3-ketoacyl-CoA thiolase, acetyl-CoA acyltransferase, or beta-ketothiolase is an enzyme that in humans is encoded by the HADHB gene.

Glycerol-3-phosphate dehydrogenase (GPDH) is an enzyme that catalyzes the reversible redox conversion of dihydroxyacetone phosphate to sn-glycerol 3-phosphate.

Potassium voltage-gated channel subfamily E member 2 (KCNE2), also known as MinK-related peptide 1 (MiRP1), is a protein that in humans is encoded by the KCNE2 gene on chromosome 21. MiRP1 is a voltage-gated potassium channel accessory subunit associated with Long QT syndrome. It is ubiquitously expressed in many tissues and cell types. Because of this and its ability to regulate multiple different ion channels, KCNE2 exerts considerable influence on a number of cell types and tissues. Human KCNE2 is a member of the five-strong family of human KCNE genes. KCNE proteins contain a single membrane-spanning region, extracellular N-terminal and intracellular C-terminal. KCNE proteins have been widely studied for their roles in the heart and in genetic predisposition to inherited cardiac arrhythmias. The KCNE2 gene also contains one of 27 SNPs associated with increased risk of coronary artery disease. More recently, roles for KCNE proteins in a variety of non-cardiac tissues have also been explored.
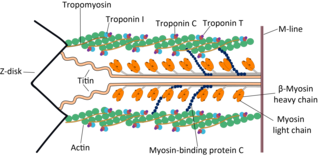
Cardiac muscle troponin T (cTnT) is a protein that in humans is encoded by the TNNT2 gene. Cardiac TnT is the tropomyosin-binding subunit of the troponin complex, which is located on the thin filament of striated muscles and regulates muscle contraction in response to alterations in intracellular calcium ion concentration.

MT-ND6 is a gene of the mitochondrial genome coding for the NADH-ubiquinone oxidoreductase chain 6 protein (ND6). The ND6 protein is a subunit of NADH dehydrogenase (ubiquinone), which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain. Variations in the human MT-ND6 gene are associated with Leigh's syndrome, Leber's hereditary optic neuropathy (LHON) and dystonia.
MT-ND2 is a gene of the mitochondrial genome coding for the NADH dehydrogenase 2 (ND2) protein. The ND2 protein is a subunit of NADH dehydrogenase (ubiquinone), which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain. Variants of human MT-ND2 are associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS), Leigh's syndrome (LS), Leber's hereditary optic neuropathy (LHON) and increases in adult BMI.
MT-ND5 is a gene of the mitochondrial genome coding for the NADH-ubiquinone oxidoreductase chain 5 protein (ND5). The ND5 protein is a subunit of NADH dehydrogenase (ubiquinone), which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain. Variations in human MT-ND5 are associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) as well as some symptoms of Leigh's syndrome and Leber's hereditary optic neuropathy (LHON).
MT-ND1 is a gene of the mitochondrial genome coding for the NADH-ubiquinone oxidoreductase chain 1 (ND1) protein. The ND1 protein is a subunit of NADH dehydrogenase, which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain. Variants of the human MT-ND1 gene are associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS), Leigh's syndrome (LS), Leber's hereditary optic neuropathy (LHON) and increases in adult BMI.

Myosin heavy chain, α isoform (MHC-α) is a protein that in humans is encoded by the MYH6 gene. This isoform is distinct from the ventricular/slow myosin heavy chain isoform, MYH7, referred to as MHC-β. MHC-α isoform is expressed predominantly in human cardiac atria, exhibiting only minor expression in human cardiac ventricles. It is the major protein comprising the cardiac muscle thick filament, and functions in cardiac muscle contraction. Mutations in MYH6 have been associated with late-onset hypertrophic cardiomyopathy, atrial septal defects and sick sinus syndrome.

Plakophilin-2 is a protein that in humans is encoded by the PKP2 gene. Plakophilin 2 is expressed in skin and cardiac muscle, where it functions to link cadherins to intermediate filaments in the cytoskeleton. In cardiac muscle, plakophilin-2 is found in desmosome structures located within intercalated discs. Mutations in PKP2 have been shown to be causal in arrhythmogenic right ventricular cardiomyopathy.
NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 9 is an enzyme that in humans is encoded by the NDUFA9 gene. The NDUFA9 protein is a subunit of NADH:ubiquinone oxidoreductase, which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain. Mutations in NADH dehydrogenase (ubiquinone), also known as Complex I, frequently lead to complex neurodegenerative diseases such as Leigh's syndrome. In the case of NDUFA9, a mutation to the MT-ND3 gene might interrupt their interaction and formation of subcomplexes, compromising Complex I function and leading to disease.
Ankyrin-2, also known as Ankyrin-B, and Brain ankyrin, is a protein which in humans is encoded by the ANK2 gene. Ankyrin-2 is ubiquitously expressed, but shows high expression in cardiac muscle. Ankyrin-2 plays an essential role in the localization and membrane stabilization of ion transporters and ion channels in cardiomyocytes, as well as in costamere structures. Mutations in ANK2 cause a dominantly-inherited, cardiac arrhythmia syndrome known as long QT syndrome 4 as well as sick sinus syndrome; mutations have also been associated to a lesser degree with hypertrophic cardiomyopathy. Alterations in ankyrin-2 expression levels are observed in human heart failure.

KCNE1-like also known as KCNE1L is a protein that in humans is encoded by the KCNE1L gene.

It remains a difficult medical challenge to prevent the sudden cardiac death of athletes, typically defined as natural, unexpected death from cardiac arrest within one hour of the onset of collapse symptoms, excluding additional time on mechanical life support. Most causes relate to congenital or acquired cardiovascular disease with no symptoms noted before the fatal event. The prevalence of any single, associated condition is low, probably less than 0.3% of the population in the athletes' age group, and the sensitivity and specificity of common screening tests leave much to be desired. The single most important predictor is fainting or near-fainting during exercise, which should require detailed explanation and investigation. The victims include many well-known names, especially in professional soccer, and close relatives are often at risk for similar cardiac problems.
Glycerol-3-phosphate dehydrogenase (EC 1.1.5.3 is an enzyme with systematic name sn-glycerol 3-phosphate:quinone oxidoreductase. This enzyme catalyses the following chemical reaction
Glycerol-3-phosphate dehydrogenase 1 is a protein that in humans is encoded by the GPD1 gene.
References
- 1 2 3 Nagase T, Miyajima N, Tanaka A, Sazuka T, Seki N, Sato S, Tabata S, Ishikawa K, Kawarabayasi Y, Kotani H (1995). "Prediction of the coding sequences of unidentified human genes. III. The coding sequences of 40 new genes (KIAA0081-KIAA0120) deduced by analysis of cDNA clones from human cell line KG-1". DNA Res. 2 (1): 37–43. doi: 10.1093/dnares/2.1.37 . PMID 7788527.
- 1 2 London B, Michalec M, Mehdi H, Zhu X, Kerchner L, Sanyal S, Viswanathan PC, Pfahnl AE, Shang LL, Madhusudanan M, Baty CJ, Lagana S, Aleong R, Gutmann R, Ackerman MJ, McNamara DM, Weiss R, Dudley SC Jr (November 2007). "Mutation in Glycerol-3-Phosphate Dehydrogenase 1–Like Gene (GPD1-L) Decreases Cardiac Na+ Current and Causes Inherited Arrhythmias". Circulation. 116 (20): 2260–8. doi:10.1161/CIRCULATIONAHA.107.703330. PMC 3150966 . PMID 17967977.
- ↑ Van Norstrand DW, Valdivia CR, Tester DJ, Ueda K, London B, Makielski JC, Ackerman MJ (November 2007). "Molecular and functional characterization of novel glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) mutations in sudden infant death syndrome". Circulation. 116 (20): 2253–9. doi:10.1161/CIRCULATIONAHA.107.704627. PMC 3332545 . PMID 17967976.