
In biology, a mutation is an alteration in the nucleic acid sequence of the genome of an organism, virus, or extrachromosomal DNA. Viral genomes contain either DNA or RNA. Mutations result from errors during DNA or viral replication, mitosis, or meiosis or other types of damage to DNA, which then may undergo error-prone repair, cause an error during other forms of repair, or cause an error during replication. Mutations may also result from substitution, insertion or deletion of segments of DNA due to mobile genetic elements.

Pelizaeus–Merzbacher disease is an X-linked neurological disorder that damages oligodendrocytes in the central nervous system. It is caused by mutations in proteolipid protein 1 (PLP1), a major myelin protein. It is characterized by a decrease in the amount of insulating myelin surrounding the nerves (hypomyelination) and belongs to a group of genetic diseases referred to as leukodystrophies.

Limb–girdle muscular dystrophy (LGMD) is a genetically heterogeneous group of rare muscular dystrophies that share a set of clinical characteristics. It is characterised by progressive muscle wasting which affects predominantly hip and shoulder muscles. LGMD usually has an autosomal pattern of inheritance. It currently has no known cure or treatment.

Harlequin-type ichthyosis is a genetic disorder that results in thickened skin over nearly the entire body at birth. The skin forms large, diamond/trapezoid/rectangle-shaped plates that are separated by deep cracks. These affect the shape of the eyelids, nose, mouth, and ears and limit movement of the arms and legs. Restricted movement of the chest can lead to breathing difficulties. These plates fall off over several weeks. Other complications can include premature birth, infection, problems with body temperature, and dehydration. The condition is the most severe form of ichthyosis, a group of genetic disorders characterised by scaly skin.

In genetics and bioinformatics, a single-nucleotide polymorphism is a germline substitution of a single nucleotide at a specific position in the genome. Although certain definitions require the substitution to be present in a sufficiently large fraction of the population, many publications do not apply such a frequency threshold.
Miller–Dieker syndrome, also called Miller–Dieker lissencephaly syndrome (MDLS) or chromosome 17p13.3 deletion syndrome, is a micro deletion syndrome characterized by congenital malformations. Congenital malformations are physical defects detectable in an infant at birth which can involve many different parts of the body, including the brain, heart, lungs, liver, bones, or intestinal tract. MDS is a contiguous gene syndrome – a disorder due to the deletion of multiple gene loci adjacent to one another. The disorder arises from the deletion of part of the small arm of chromosome 17p, leading to partial monosomy. There may be unbalanced translocations, or the presence of a ring chromosome 17.
Phakomatoses, also known as neurocutaneous syndromes, are a group of multisystemic diseases that most prominently affect structures primarily derived from the ectoderm such as the central nervous system, skin and eyes. The majority of phakomatoses are single-gene disorders that may be inherited in an autosomal dominant, autosomal recessive or X-linked pattern. Presentations may vary dramatically between patients with the same particular syndrome due to mosaicism, variable expressivity, and penetrance.

T-box transcription factor T, also known as Brachyury protein, is encoded for in humans and other apes by the TBXT gene. Brachyury functions as a transcription factor within the T-box family of genes. Brachyury homologs have been found in all bilaterian animals that have been screened, as well as the freshwater cnidarian Hydra.
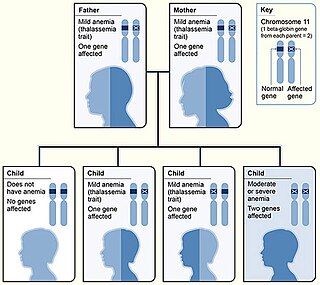
Beta thalassemias are a group of inherited blood disorders. They are forms of thalassemia caused by reduced or absent synthesis of the beta chains of hemoglobin that result in variable outcomes ranging from severe anemia to clinically asymptomatic individuals. Global annual incidence is estimated at one in 100,000. Beta thalassemias occur due to malfunctions in the hemoglobin subunit beta or HBB. The severity of the disease depends on the nature of the mutation.

A dysmorphic feature is an abnormal difference in body structure. It can be an isolated finding in an otherwise normal individual, or it can be related to a congenital disorder, genetic syndrome or birth defect. Dysmorphology is the study of dysmorphic features, their origins and proper nomenclature. One of the key challenges in identifying and describing dysmorphic features is the use and understanding of specific terms between different individuals. Clinical geneticists and pediatricians are usually those most closely involved with the identification and description of dysmorphic features, as most are apparent during childhood.

Whole genome sequencing (WGS) is the process of determining the entirety, or nearly the entirety, of the DNA sequence of an organism's genome at a single time. This entails sequencing all of an organism's chromosomal DNA as well as DNA contained in the mitochondria and, for plants, in the chloroplast.
1q21.1 deletion syndrome is a rare aberration of chromosome 1. A human cell has one pair of identical chromosomes on chromosome 1. With the 1q21.1 deletion syndrome, one chromosome of the pair is not complete, because a part of the sequence of the chromosome is missing. One chromosome has the normal length and the other is too short.

1q21.1 duplication syndrome, also known as 1q21.1 microduplication, is an uncommon copy number variant associated with several congenital abnormalities, including developmental delay, dysmorphic traits, autism spectrum disorder, and congenital cardiac defects. Common facial features include frontal bossing, hypertelorism, and macrocephaly. Around 18 and 29% of patients with 1q21.1 microduplications have congenital cardiac abnormalities. 1q21.1 duplication syndrome is caused by microduplications of the BP3-BP4 region. 18-50% are de novo deletions and 50-82% inherited from parents. The 1q21.1 area, one of the largest regions in the human genome, is highly susceptible to copy number variation due to its frequent low-copy duplications. Whole exon sequencing and quantitative polymerase chain reaction can provide a precise molecular diagnosis for children with 1q21.1 microduplication syndrome.

Hyperphosphatasia with mental retardation syndrome, HPMRS, also known as Mabry syndrome, has been described in patients recruited on four continents world-wide. Mabry syndrome was confirmed to represent an autosomal recessive syndrome characterized by severe mental retardation, considerably elevated serum levels of alkaline phosphatase, hypoplastic terminal phalanges, and distinct facial features that include: hypertelorism, a broad nasal bridge and a rectangular face.
Single nucleotide polymorphism annotation is the process of predicting the effect or function of an individual SNP using SNP annotation tools. In SNP annotation the biological information is extracted, collected and displayed in a clear form amenable to query. SNP functional annotation is typically performed based on the available information on nucleic acid and protein sequences.

A topologically associating domain (TAD) is a self-interacting genomic region, meaning that DNA sequences within a TAD physically interact with each other more frequently than with sequences outside the TAD. The average size of a topologically associating domain (TAD) is 1000 kb in humans, 880 kb in mouse cells, and 140 kb in fruit flies. Boundaries at both side of these domains are conserved between different mammalian cell types and even across species and are highly enriched with CCCTC-binding factor (CTCF) and cohesin. In addition, some types of genes appear near TAD boundaries more often than would be expected by chance.

Alcohol intolerance is due to a genetic polymorphism of the aldehyde dehydrogenase enzyme, which is responsible for the metabolism of acetaldehyde. This polymorphism is most often reported in patients of East Asian descent. Alcohol intolerance may also be an associated side effect of certain drugs such as disulfiram, metronidazole, or nilutamide. Skin flushing and nasal congestion are the most common symptoms of intolerance after alcohol ingestion. It may also be characterized as intolerance causing hangover symptoms similar to the "disulfiram-like reaction" of aldehyde dehydrogenase deficiency or chronic fatigue syndrome. Severe pain after drinking alcohol may indicate a more serious underlying condition.

In genetics, a polygenic score (PGS) is a number that summarizes the estimated effect of many genetic variants on an individual's phenotype. The PGS is also called the polygenic index (PGI) or genome-wide score; in the context of disease risk, it is called a polygenic risk score or genetic risk score. The score reflects an individual's estimated genetic predisposition for a given trait and can be used as a predictor for that trait. It gives an estimate of how likely an individual is to have a given trait based only on genetics, without taking environmental factors into account; and it is typically calculated as a weighted sum of trait-associated alleles.
Bainbridge–Ropers syndrome was first identified in 2013 and is characterized by failure to thrive, feeding problems, hypotonia, intellectual disabilities, autism, postnatal growth delay, abnormal facial features such as arched eyebrows, anteverted nares, and delays in language acquisition. BRPS is extremely rare worldwide; more than thirty cases of BRPS have been reported abroad, and four cases have been reported in China.
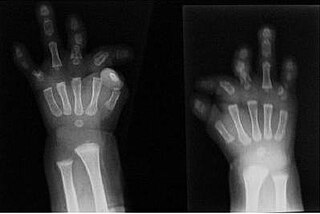
Du Pan syndrome, also known as fibular aplasia-complex brachydactyly syndrome, is an extremely rare genetic condition. Unlike other rare genetic conditions, Du Pan syndrome does not affect brain function or the appearance of the head and trunk. This condition is associated with alterations to the GDF5 gene. The way that this condition is passed on from generation to generation varies, but it is most commonly inherited in an autosomal recessive manner, meaning two copies of the same version of the gene are required to show this condition. Rare cases exist where the mode of inheritance is autosomal dominant, which means having only one version of the gene is enough to cause this condition.
