Related Research Articles

Protein secondary structure is the local spatial conformation of the polypeptide backbone excluding the side chains. The two most common secondary structural elements are alpha helices and beta sheets, though beta turns and omega loops occur as well. Secondary structure elements typically spontaneously form as an intermediate before the protein folds into its three dimensional tertiary structure.

Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design.
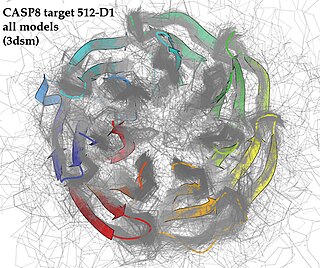
Critical Assessment of Structure Prediction (CASP), sometimes called Critical Assessment of Protein Structure Prediction, is a community-wide, worldwide experiment for protein structure prediction taking place every two years since 1994. CASP provides research groups with an opportunity to objectively test their structure prediction methods and delivers an independent assessment of the state of the art in protein structure modeling to the research community and software users. Even though the primary goal of CASP is to help advance the methods of identifying protein three-dimensional structure from its amino acid sequence many view the experiment more as a "world championship" in this field of science. More than 100 research groups from all over the world participate in CASP on a regular basis and it is not uncommon for entire groups to suspend their other research for months while they focus on getting their servers ready for the experiment and on performing the detailed predictions.

Structural alignment attempts to establish homology between two or more polymer structures based on their shape and three-dimensional conformation. This process is usually applied to protein tertiary structures but can also be used for large RNA molecules. In contrast to simple structural superposition, where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment techniques. Structural alignment can therefore be used to imply evolutionary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution by which multiple unrelated amino acid sequences converge on a common tertiary structure.

Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. The main objective of structural bioinformatics is the creation of new methods of analysing and manipulating biological macromolecular data in order to solve problems in biology and generate new knowledge.
In molecular biology, protein threading, also known as fold recognition, is a method of protein modeling which is used to model those proteins which have the same fold as proteins of known structures, but do not have homologous proteins with known structure. It differs from the homology modeling method of structure prediction as it is used for proteins which do not have their homologous protein structures deposited in the Protein Data Bank (PDB), whereas homology modeling is used for those proteins which do. Threading works by using statistical knowledge of the relationship between the structures deposited in the PDB and the sequence of the protein which one wishes to model.
Rosetta@home is a volunteer computing project researching protein structure prediction on the Berkeley Open Infrastructure for Network Computing (BOINC) platform, run by the Baker lab. Rosetta@home aims to predict protein–protein docking and design new proteins with the help of about fifty-five thousand active volunteered computers processing at over 487,946 GigaFLOPS on average as of September 19, 2020. Foldit, a Rosetta@home videogame, aims to reach these goals with a crowdsourcing approach. Though much of the project is oriented toward basic research to improve the accuracy and robustness of proteomics methods, Rosetta@home also does applied research on malaria, Alzheimer's disease, and other pathologies.
In bioinformatics, the root mean square deviation of atomic positions, or simply root mean square deviation (RMSD), is the measure of the average distance between the atoms (usually the backbone atoms) of superimposed molecules. In the study of globular protein conformations, one customarily measures the similarity in three-dimensional structure by the RMSD of the Cα atomic coordinates after optimal rigid body superposition.

Homology modeling, also known as comparative modeling of protein, refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental three-dimensional structure of a related homologous protein. Homology modeling relies on the identification of one or more known protein structures likely to resemble the structure of the query sequence, and on the production of a sequence alignment that maps residues in the query sequence to residues in the template sequence. It has been seen that protein structures are more conserved than protein sequences amongst homologues, but sequences falling below a 20% sequence identity can have very different structure.
In computational biology, de novo protein structure prediction refers to an algorithmic process by which protein tertiary structure is predicted from its amino acid primary sequence. The problem itself has occupied leading scientists for decades while still remaining unsolved. According to Science, the problem remains one of the top 125 outstanding issues in modern science. At present, some of the most successful methods have a reasonable probability of predicting the folds of small, single-domain proteins within 1.5 angstroms over the entire structure.
RAPTOR is protein threading software used for protein structure prediction. It has been replaced by RaptorX, which is much more accurate than RAPTOR.
In bioinformatics, the template modeling score or TM-score is a measure of similarity between two protein structures. The TM-score is intended as a more accurate measure of the global similarity of full-length protein structures than the often used RMSD measure. The TM-score indicates the similarity between two structures by a score between , where 1 indicates a perfect match between two structures. Generally scores below 0.20 corresponds to randomly chosen unrelated proteins whereas structures with a score higher than 0.5 assume roughly the same fold. A quantitative study shows that proteins of TM-score = 0.5 have a posterior probability of 37% in the same CATH topology family and of 13% in the same SCOP fold family. The probabilities increase rapidly when TM-score > 0.5. The TM-score is designed to be independent of protein lengths.
Phyre and Phyre2 are free web-based services for protein structure prediction. Phyre is among the most popular methods for protein structure prediction having been cited over 1,500 times. Like other remote homology recognition techniques, it is able to regularly generate reliable protein models when other widely used methods such as PSI-BLAST cannot. Phyre2 has been designed to ensure a user-friendly interface for users inexpert in protein structure prediction methods. Its development is funded by the Biotechnology and Biological Sciences Research Council.
RaptorX is a software and web server for protein structure and function prediction that is free for non-commercial use. RaptorX is among the most popular methods for protein structure prediction. Like other remote homology recognition and protein threading techniques, RaptorX is able to regularly generate reliable protein models when the widely used PSI-BLAST cannot. However, RaptorX is also significantly different from profile-based methods in that RaptorX excels at modeling of protein sequences without a large number of sequence homologs by exploiting structure information. RaptorX Server has been designed to ensure a user-friendly interface for users inexpert in protein structure prediction methods.
Swiss-model is a structural bioinformatics web-server dedicated to homology modeling of 3D protein structures. As of 2024, homology modeling is the most accurate method to generate reliable three-dimensional protein structure models and is routinely used in many practical applications. Homology modelling methods make use of experimental protein structures (templates) to build models for evolutionary related proteins (targets).
Continuous Automated Model EvaluatiOn (CAMEO) is a community-wide project to continuously evaluate the accuracy and reliability of protein structure prediction servers in a fully automated manner. CAMEO is a continuous and fully automated complement to the bi-annual CASP experiment.

I-TASSER is a bioinformatics method for predicting three-dimensional structure model of protein molecules from amino acid sequences. It detects structure templates from the Protein Data Bank by a technique called fold recognition. The full-length structure models are constructed by reassembling structural fragments from threading templates using replica exchange Monte Carlo simulations. I-TASSER is one of the most successful protein structure prediction methods in the community-wide CASP experiments.
AlphaFold is an artificial intelligence (AI) program developed by DeepMind, a subsidiary of Alphabet, which performs predictions of protein structure. The program is designed as a deep learning system.
IntFOLD is fully automated, integrated pipeline for prediction of 3D structure and function from amino acid sequences. The pipeline is wrapped up and deployed as a Web Server. The core of the server method is quality assessment using built-in accuracy self-estimates (ASE) which improves performance prediction of 3D model using ModFOLD.

John Michael Jumper is an American chemist and computer scientist. He currently serves as director at Google DeepMind. Jumper and his colleagues created AlphaFold, an artificial intelligence (AI) model to predict protein structures from their amino acid sequence with high accuracy. Jumper stated that the AlphaFold team plans to release 100 million protein structures.
References
- 1 2 3 4 5 6 Zemla A (2003). "LGA: A method for finding 3D similarities in protein structures". Nucleic Acids Research. 31 (13): 3370–3374. doi:10.1093/nar/gkg571. PMC 168977 . PMID 12824330.
- 1 2 USpatent 8024127 B2,Adam Zemla,"Local-Global Alignment for Finding 3D Similarities in Protein Structures",issued 20 September 2011, assigned to Lawrence Livermore National Security, LLC
- 1 2 Zemla A, Venclovas C, Moult J, Fidelis K (1999). "Processing and analysis of CASP3 protein structure predictions". Proteins. S3 (S3): 22–29. doi:10.1002/(SICI)1097-0134(1999)37:3+<22::AID-PROT5>3.0.CO;2-W. PMID 10526349. S2CID 29803757.
- ↑ Zemla A, Venclovas C, Moult J, Fidelis K (2001). "Processing and evaluation of predictions in CASP4". Proteins. 45 (S5): 13–21. doi:10.1002/prot.10052. PMID 11835478. S2CID 28166260.
- 1 2 3 Keedy, D.A.; Williams, CJ; Headd, JJ; Arendall, WB; Chen, VB; Kapral, GJ; Gillespie, RA; Block, JN; Zemla, A; Richardson, DC; Richardson, JS (2009). "The other 90% of the protein: Assessment beyond the α-carbon for CASP8 template-based and high-accuracy models". Proteins. 77 (Suppl 9): 29–49. doi:10.1002/prot.22551. PMC 2877634 . PMID 19731372.
- ↑ Kryshtafovych, A; Prlic, A; Dmytriv, Z; Daniluk, P; Milostan, M; Eyrich, V; Hubbard, T; Fidelis, K (2007). "New tools and expanded data analysis capabilities at the Protein Structure Prediction Center". Proteins. 69 Suppl 8 (S8): 19–26. doi:10.1002/prot.21653. PMC 2656758 . PMID 17705273.
- 1 2 "Results Table Help". 14th Community Wide Experiment on the Critical Assessment of Techniques for Protein Structure Prediction. Retrieved 27 December 2020.
- ↑ Read, Randy J.; Chavali, Gayatri (2007). "Assessment of CASP7 predictions in the high accuracy template-based modeling category". Proteins. 69 (S8): 27–37. doi: 10.1002/prot.21662 . PMID 17894351. S2CID 33172629.
- ↑ Shi, S; Pei, J; Sadreyev, RI; Kinch, LN; Majumdar, I; Tong, J; Cheng, H; Kim, BH; Grishin, NV (2009). "Analysis of CASP8 targets, predictions and assessment methods". Database: The Journal of Biological Databases and Curation. 2009: bap003. doi:10.1093/database/bap003. PMC 2794793 . PMID 20157476.. Related page
- ↑ Sadreyev, RI; Shi, S; Baker, D; Grishin, NV (15 May 2009). "Structure similarity measure with penalty for close non-equivalent residues". Bioinformatics. 25 (10): 1259–63. doi:10.1093/bioinformatics/btp148. PMC 2677741 . PMID 19321733.
- ↑ Modi V, Xu QF, Adhikari S, Dunbrack RL (2016). "Assessment of template-based modeling of protein structure in CASP11". Proteins. 84 (Suppl 1): 200–220. doi:10.1002/prot.25049. PMC 5030193 . PMID 27081927.
- ↑ MacCallum, Justin L.; Hua, Lan; Schnieders, Michael J.; Pande, Vijay S.; Jacobson, Matthew P.; Dill, Ken A. (2009). "Assessment of the protein-structure refinement category in CASP8". Proteins: Structure, Function, and Bioinformatics. 77 (S9): 66–80. doi: 10.1002/prot.22538 . PMC 2801025 . PMID 19714776.
- ↑ Li, Wenlin; Schaeffer, R. Dustin; Otwinowski, Zbyszek; Grishin, Nick V. (5 May 2016). "Estimation of Uncertainties in the Global Distance Test (GDT_TS) for CASP Models". PLOS ONE. 11 (5): e0154786. Bibcode:2016PLoSO..1154786L. doi: 10.1371/journal.pone.0154786 . PMC 4858170 . PMID 27149620.