Related Research Articles

Bioinformatics is an interdisciplinary field that develops methods and software tools for understanding biological data, in particular when the data sets are large and complex. As an interdisciplinary field of science, bioinformatics combines biology, computer science, information engineering, mathematics and statistics to analyze and interpret the biological data. Bioinformatics has been used for in silico analyses of biological queries using mathematical and statistical techniques.

The proteome is the entire set of proteins that is, or can be, expressed by a genome, cell, tissue, or organism at a certain time. It is the set of expressed proteins in a given type of cell or organism, at a given time, under defined conditions. Proteomics is the study of the proteome.

Proteomics is the large-scale study of proteins. Proteins are vital parts of living organisms, with many functions. The word proteome is a portmanteau of protein and genome, and was coined by Marc Wilkins in 1994 while he was a Ph.D. student at Macquarie University. Macquarie University also founded the first dedicated proteomics laboratory in 1995.

Peptide mass fingerprinting (PMF) is an analytical technique for protein identification in which the unknown protein of interest is first cleaved into smaller peptides, whose absolute masses can be accurately measured with a mass spectrometer such as MALDI-TOF or ESI-TOF. The method was developed in 1993 by several groups independently. The peptide masses are compared to either a database containing known protein sequences or even the genome. This is achieved by using computer programs that translate the known genome of the organism into proteins, then theoretically cut the proteins into peptides, and calculate the absolute masses of the peptides from each protein. They then compare the masses of the peptides of the unknown protein to the theoretical peptide masses of each protein encoded in the genome. The results are statistically analyzed to find the best match.

Stable Isotope Labeling by/with Amino acids in Cell culture (SILAC) is a technique based on mass spectrometry that detects differences in protein abundance among samples using non-radioactive isotopic labeling. It is a popular method for quantitative proteomics.
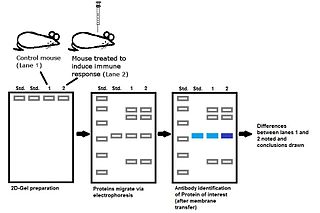
Immunoproteomics is the study of large sets of proteins (proteomics) involved in the immune response.
The Trans-Proteomic Pipeline (TPP) is an open-source data analysis software for proteomics developed at the Institute for Systems Biology (ISB) by the Ruedi Aebersold group under the Seattle Proteome Center. The TPP includes PeptideProphet, ProteinProphet, ASAPRatio, XPRESS and Libra.
Surface-enhanced laser desorption/ionization (SELDI) is a soft ionization method in mass spectrometry (MS) used for the analysis of protein mixtures. It is a variation of matrix-assisted laser desorption/ionization (MALDI). In MALDI, the sample is mixed with a matrix material and applied to a metal plate before irradiation by a laser, whereas in SELDI, proteins of interest in a sample become bound to a surface before MS analysis. The sample surface is a key component in the purification, desorption, and ionization of the sample. SELDI is typically used with time-of-flight (TOF) mass spectrometers and is used to detect proteins in tissue samples, blood, urine, or other clinical samples, however, SELDI technology can potentially be used in any application by simply modifying the sample surface.
Shotgun proteomics refers to the use of bottom-up proteomics techniques in identifying proteins in complex mixtures using a combination of high performance liquid chromatography combined with mass spectrometry. The name is derived from shotgun sequencing of DNA which is itself named after the rapidly expanding, quasi-random firing pattern of a shotgun. The most common method of shotgun proteomics starts with the proteins in the mixture being digested and the resulting peptides are separated by liquid chromatography. Tandem mass spectrometry is then used to identify the peptides.

Top-down proteomics is a method of protein identification that either uses an ion trapping mass spectrometer to store an isolated protein ion for mass measurement and tandem mass spectrometry (MS/MS) analysis or other protein purification methods such as two-dimensional gel electrophoresis in conjunction with MS/MS. Top-down proteomics is capable of identifying and quantitating unique proteoforms through the analysis of intact proteins. The name is derived from the similar approach to DNA sequencing. During mass spectrometry intact proteins are typically ionized by electrospray ionization and trapped in a Fourier transform ion cyclotron resonance, quadrupole ion trap or Orbitrap mass spectrometer. Fragmentation for tandem mass spectrometry is accomplished by electron-capture dissociation or electron-transfer dissociation. Effective fractionation is critical for sample handling before mass-spectrometry-based proteomics. Proteome analysis routinely involves digesting intact proteins followed by inferred protein identification using mass spectrometry (MS). Top-down MS (non-gel) proteomics interrogates protein structure through measurement of an intact mass followed by direct ion dissociation in the gas phase.

Isobaric tags for relative and absolute quantitation (iTRAQ) is an isobaric labeling method used in quantitative proteomics by tandem mass spectrometry to determine the amount of proteins from different sources in a single experiment. It uses stable isotope labeled molecules that can be covalent bonded to the N-terminus and side chain amines of proteins.
The Proteomics Standards Initiative (PSI) is a working group of Human Proteome Organization. It aims to define data standards for proteomics in order to facilitate data comparison, exchange and verification.
Edward Marcotte is a professor of biochemistry at The University of Texas at Austin, working in genetics, proteomics, and bioinformatics. Marcotte is an example of a computational biologist who also relies on experiments to validate bioinformatics-based predictions.

Isobaric labeling is a mass spectrometry strategy used in quantitative proteomics. Peptides or proteins are labeled with various chemical groups that are identical masses (isobaric), but vary in terms of distribution of heavy isotopes around their structure. These tags, commonly referred to as tandem mass tags, are designed so that the mass tag is cleaved at a specific linker region upon high-energy CID (HCD) during tandem mass spectrometry yielding reporter ions of different masses. The most common isobaric tags are amine-reactive tags. However, tags that react with cysteine residues and carbonyl groups have also been described. These amine-reactive groups go through N-hydroxysuccinimide (NHS) reactions, which are based around three types of functional groups. Isobaric labeling methods include tandem mass tags (TMT), isobaric tags for absolute and relative quantification (iTRAQ), mass differential tags for absolute and relative quantification, and dimethyl labeling. TMTs and iTRAQ methods are most common and developed of these methods. Tandem mass tags have a mass reporter region, a cleavable linker region, a mass normalization region, and a protein reactive group and have the same total mass.
In the field of cellular biology, single-cell analysis is the study of genomics, transcriptomics, proteomics and metabolomics at the single cell level. Due to the heterogeneity seen in both eukaryotic and prokaryotic cell populations, analyzing a single cell makes it possible to discover mechanisms not seen when studying a bulk population of cells. Technologies such as fluorescence-activated cell sorting (FACS) allow the precise isolation of selected single cells from complex samples, while high throughput single cell partitioning technologies, enable the simultaneous molecular analysis of hundreds or thousands of single unsorted cells; this is particularly useful for the analysis of transcriptome variation in genotypically identical cells, allowing the definition of otherwise undetectable cell subtypes. The development of new technologies is increasing our ability to analyze the genome, and transcriptome, of single cells, as well as to quantify their proteome and metabolome. New developments in mass spectrometry techniques have become important analytical tools for proteomic and metabolomic analysis of single cells. In situ sequencing and fluorescence in situ hybridization (FISH) do not require that cells be isolated and are increasingly being used for analysis of tissues.
MassMatrix is a mass spectrometry data analysis software that uses a statistical model to achieve increased mass accuracy over other database search algorithms. This search engine is set apart from others dues to its ability to provide extremely efficient judgement between true and false positives for high mass accuracy data that has been obtained from present day mass spectrometer instruments. It is useful for identifying disulphide bonds in tandem mass spectrometry data. This search engine is set apart from others due to its ability to provide extremely efficient judgement between true and false positives for high mass accuracy data that has been obtained from present day mass spectrometer instruments.

Ronald Charles Beavis is a Canadian protein biochemist, who has been involved in the application of mass spectrometry to protein primary structure, with applications in the fields of proteomics and analytical biochemistry. He has developed methods for measuring the identity and post-translational modification state of proteins obtained from biological samples using mass spectrometry. He is currently best known for developing new methods for analyzing proteomics data and applying the results of these methods to problems in computational biology.
Unipept is an open source research tool for biodiversity analysis of metaproteomics samples. It also contains a tool to select peptides to use as biomarker and a tool to compare the genome of organisms based on their protein content. The software is developed at Ghent University.
David Fenyö is a Professor in the Department of Biochemistry and Molecular Pharmacology at NYU Langone Medical Center, and the Director for the Ph.D. program in biomedical informatics. David heads the NYU Laboratory of Computational Proteomics focusing on the development of methods to identify, characterize and quantify proteins and in the integration of data from multiple modalities including mass spectrometry, sequencing and microscopy.
References
- ↑ Craig, Robertson; Cortens, John P.; Beavis, Ronald C. (2004). "Open Source System for Analyzing, Validating, and Storing Protein Identification Data". Journal of Proteome Research. 3 (6): 1234–42. doi:10.1021/pr049882h. PMID 15595733.
- ↑ Fenyo, D (1999). "The Biopolymer Markup Language". Bioinformatics. 15 (4): 339–40. doi: 10.1093/bioinformatics/15.4.339 . PMID 10320402.
- ↑ Craig, Robertson; Cortens, John P.; Beavis, Ronald C. (2004). "Open Source System for Analyzing, Validating, and Storing Protein Identification Data". Journal of Proteome Research. 3 (6): 1234–1242. doi:10.1021/pr049882h. ISSN 1535-3893. PMID 15595733.
- ↑ Omenn, Gilbert S.; Lane, Lydie; Lundberg, Emma K.; Beavis, Ronald C.; Nesvizhskii, Alexey I.; Deutsch, Eric W. (2015). "Metrics for the Human Proteome Project 2015: Progress on the Human Proteome and Guidelines for High-Confidence Protein Identification". Journal of Proteome Research. 14 (9): 3452–3460. doi:10.1021/acs.jproteome.5b00499. ISSN 1535-3893. PMC 4755311 . PMID 26155816.
- ↑ Fenyö, David; Beavis, Ronald C. (2015). "The GPMDB REST interface". Bioinformatics. 31 (12): 2056–2058. doi: 10.1093/bioinformatics/btv107 . ISSN 1367-4803. PMID 25697819.