
In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns. Sequence alignments are also used for non-biological sequences such as calculating the distance cost between strings in a natural language, or to display financial data.
Protein engineering is the process of developing useful or valuable proteins through the design and production of unnatural polypeptides, often by altering amino acid sequences found in nature. It is a young discipline, with much research taking place into the understanding of protein folding and recognition for protein design principles. It has been used to improve the function of many enzymes for industrial catalysis. It is also a product and services market, with an estimated value of $168 billion by 2017.

Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by computational biology; it is important in medicine and biotechnology.
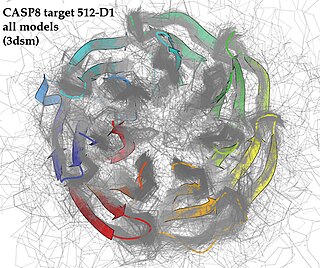
Critical Assessment of Structure Prediction (CASP), sometimes called Critical Assessment of Protein Structure Prediction, is a community-wide, worldwide experiment for protein structure prediction taking place every two years since 1994. CASP provides research groups with an opportunity to objectively test their structure prediction methods and delivers an independent assessment of the state of the art in protein structure modeling to the research community and software users. Even though the primary goal of CASP is to help advance the methods of identifying protein three-dimensional structure from its amino acid sequence many view the experiment more as a “world championship” in this field of science. More than 100 research groups from all over the world participate in CASP on a regular basis and it is not uncommon for entire groups to suspend their other research for months while they focus on getting their servers ready for the experiment and on performing the detailed predictions.

A protein family is a group of evolutionarily related proteins. In many cases, a protein family has a corresponding gene family, in which each gene encodes a corresponding protein with a 1:1 relationship. The term "protein family" should not be confused with family as it is used in taxonomy.
The European Bioinformatics Institute (EMBL-EBI) is an intergovernmental organization (IGO) which, as part of the European Molecular Biology Laboratory (EMBL) family, focuses on research and services in bioinformatics. It is located on the Wellcome Genome Campus in Hinxton near Cambridge, and employs over 600 full-time equivalent (FTE) staff. Institute leaders such as Rolf Apweiler, Alex Bateman, Ewan Birney, and Guy Cochrane, an adviser on the National Genomics Data Center Scientific Advisory Board, serve as part of the international research network of the BIG Data Center at the Beijing Institute of Genomics.
In molecular biology, protein threading, also known as fold recognition, is a method of protein modeling which is used to model those proteins which have the same fold as proteins of known structures, but do not have homologous proteins with known structure. It differs from the homology modeling method of structure prediction as it is used for proteins which do not have their homologous protein structures deposited in the Protein Data Bank (PDB), whereas homology modeling is used for those proteins which do. Threading works by using statistical knowledge of the relationship between the structures deposited in the PDB and the sequence of the protein which one wishes to model.

Multiple sequence alignment (MSA) is the process or the result of sequence alignment of three or more biological sequences, generally protein, DNA, or RNA. These alignments are used to infer evolutionary relationships via phylogenetic analysis and can highlight homologous features between sequences. Alignments highlight mutation events such as point mutations, insertion mutations and deletion mutations, and alignments are used to assess sequence conservation and infer the presence and activity of protein domains, tertiary structures, secondary structures, and individual amino acids or nucleotides.
InterPro is a database of protein families, protein domains and functional sites in which identifiable features found in known proteins can be applied to new protein sequences in order to functionally characterise them.

Homology modeling, also known as comparative modeling of protein, refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental three-dimensional structure of a related homologous protein. Homology modeling relies on the identification of one or more known protein structures likely to resemble the structure of the query sequence, and on the production of an alignment that maps residues in the query sequence to residues in the template sequence. It has been seen that protein structures are more conserved than protein sequences amongst homologues, but sequences falling below a 20% sequence identity can have very different structure.
Loop modeling is a problem in protein structure prediction requiring the prediction of the conformations of loop regions in proteins with or without the use of a structural template. Computer programs that solve these problems have been used to research a broad range of scientific topics from ADP to breast cancer. Because protein function is determined by its shape and the physiochemical properties of its exposed surface, it is important to create an accurate model for protein/ligand interaction studies. The problem arises often in homology modeling, where the tertiary structure of an amino acid sequence is predicted based on a sequence alignment to a template, or a second sequence whose structure is known. Because loops have highly variable sequences even within a given structural motif or protein fold, they often correspond to unaligned regions in sequence alignments; they also tend to be located at the solvent-exposed surface of globular proteins and thus are more conformationally flexible. Consequently, they often cannot be modeled using standard homology modeling techniques. More constrained versions of loop modeling are also used in the data fitting stages of solving a protein structure by X-ray crystallography, because loops can correspond to regions of low electron density and are therefore difficult to resolve.

HMMER is a free and commonly used software package for sequence analysis written by Sean Eddy. Its general usage is to identify homologous protein or nucleotide sequences, and to perform sequence alignments. It detects homology by comparing a profile-HMM to either a single sequence or a database of sequences. Sequences that score significantly better to the profile-HMM compared to a null model are considered to be homologous to the sequences that were used to construct the profile-HMM. Profile-HMMs are constructed from a multiple sequence alignment in the HMMER package using the hmmbuild program. The profile-HMM implementation used in the HMMER software was based on the work of Krogh and colleagues. HMMER is a console utility ported to every major operating system, including different versions of Linux, Windows, and macOS.
RAPTOR is protein threading software used for protein structure prediction. It has been replaced by RaptorX, which is much more accurate than RAPTOR.
CS-BLAST (Context-Specific BLAST) is a tool that searches a protein sequence that extends BLAST, using context-specific mutation probabilities. More specifically, CS-BLAST derives context-specific amino-acid similarities on each query sequence from short windows on the query sequences. Using CS-BLAST doubles sensitivity and significantly improves alignment quality without a loss of speed in comparison to BLAST. CSI-BLAST is the context-specific analog of PSI-BLAST, which computes the mutation profile with substitution probabilities and mixes it with the query profile. CSI-BLAST is the context specific analog of PSI-BLAST. Both of these programs are available as web-server and are available for free download.
Phyre and Phyre2 are free web-based services for protein structure prediction. Phyre is among the most popular methods for protein structure prediction having been cited over 1500 times. Like other remote homology recognition techniques, it is able to regularly generate reliable protein models when other widely used methods such as PSI-BLAST cannot. Phyre2 has been designed to ensure a user-friendly interface for users inexpert in protein structure prediction methods. Its development is funded by the Biotechnology and Biological Sciences Research Council.
RaptorX is a software and web server for protein structure and function prediction that is free for non-commercial use. RaptorX is among the most popular methods for protein structure prediction. Like other remote homology recognition/protein threading techniques, RaptorX is able to regularly generate reliable protein models when the widely used PSI-BLAST cannot. However, RaptorX is also significantly different from those profile-based methods in that RaptorX excels at modeling of protein sequences without a large number of sequence homologs by exploiting structure information. RaptorX Server has been designed to ensure a user-friendly interface for users inexpert in protein structure prediction methods.
SWISS-MODEL is a structural bioinformatics web-server dedicated to homology modeling of 3D protein structures. Homology modeling is currently the most accurate method to generate reliable three-dimensional protein structure models and is routinely used in many practical applications. Homology modelling methods make use of experimental protein structures ("templates") to build models for evolutionary related proteins ("targets").
PredictProtein (PP) is an automatic service that searches up-to-date public sequence databases, creates alignments, and predicts aspects of protein structure and function. Users send a protein sequence and receive a single file with results from database comparisons and prediction methods. PP went online in 1992 at the European Molecular Biology Laboratory; since 1999 it has operated from Columbia University and in 2009 it moved to the Technische Universität München. Although many servers have implemented particular aspects, PP remains the most widely used public server for structure prediction: over 1.5 million requests from users in 104 countries have been handled; over 13000 users submitted 10 or more different queries. PP web pages are mirrored in 17 countries on 4 continents. The system is optimized to meet the demands of experimentalists not experienced in bioinformatics. This implied that we focused on incorporating only high-quality methods, and tried to collate results omitting less reliable or less important ones.

CS23D is a web server to generate 3D structural models from NMR chemical shifts. CS23D combines maximal fragment assembly with chemical shift threading, de novo structure generation, chemical shift-based torsion angle prediction, and chemical shift refinement. CS23D makes use of RefDB and ShiftX.

I-TASSER is a bioinformatics method for predicting three-dimensional structure model of protein molecules from amino acid sequences. It detects structure templates from the Protein Data Bank by a technique called fold recognition. The full-length structure models are constructed by reassembling structural fragments from threading templates using replica exchange Monte Carlo simulations. I-TASSER is one of the most successful protein structure prediction methods in the community-wide CASP experiments.
