
An alpha helix is a sequence of amino acids in a protein that are twisted into a coil.
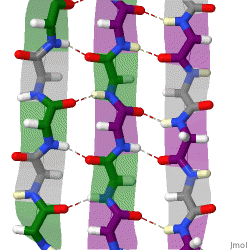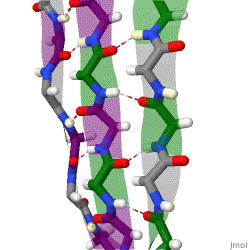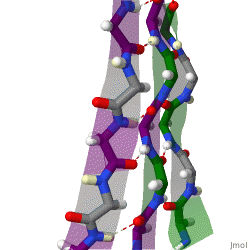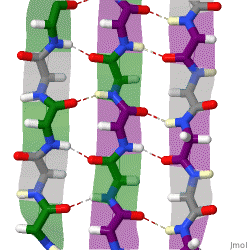
The beta sheet is a common motif of the regular protein secondary structure. Beta sheets consist of beta strands (β-strands) connected laterally by at least two or three backbone hydrogen bonds, forming a generally twisted, pleated sheet. A β-strand is a stretch of polypeptide chain typically 3 to 10 amino acids long with backbone in an extended conformation. The supramolecular association of β-sheets has been implicated in the formation of the fibrils and protein aggregates observed in amyloidosis, Alzheimer's disease and other proteinopathies.

In molecular biology, the collagen triple helix or type-2 helix is the main secondary structure of various types of fibrous collagen, including type I collagen. In 1954, Ramachandran & Kartha advanced a structure for the collagen triple helix on the basis of fiber diffraction data. It consists of a triple helix made of the repetitious amino acid sequence glycine-X-Y, where X and Y are frequently proline or hydroxyproline. Collagen folded into a triple helix is known as tropocollagen. Collagen triple helices are often bundled into fibrils which themselves form larger fibres, as in tendons.

Protein secondary structure is the local spatial conformation of the polypeptide backbone excluding the side chains. The two most common secondary structural elements are alpha helices and beta sheets, though beta turns and omega loops occur as well. Secondary structure elements typically spontaneously form as an intermediate before the protein folds into its three dimensional tertiary structure.

A transmembrane protein is a type of integral membrane protein that spans the entirety of the cell membrane. Many transmembrane proteins function as gateways to permit the transport of specific substances across the membrane. They frequently undergo significant conformational changes to move a substance through the membrane. They are usually highly hydrophobic and aggregate and precipitate in water. They require detergents or nonpolar solvents for extraction, although some of them (beta-barrels) can be also extracted using denaturing agents.

Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design.
A coiled coil is a structural motif in proteins in which 2–7 alpha-helices are coiled together like the strands of a rope. They have been found in roughly 5-10% of proteins and have a variety of functions. They are one of the most widespread motifs found in protein-protein interactions. To aid protein study, several tools have been developed to predict coiled-coils in protein structures. Many coiled coil-type proteins are involved in important biological functions, such as the regulation of gene expression — e.g., transcription factors. Notable examples are the oncoproteins c-Fos and c-Jun, as well as the muscle protein tropomyosin.

A leucine zipper is a common three-dimensional structural motif in proteins. They were first described by Landschulz and collaborators in 1988 when they found that an enhancer binding protein had a very characteristic 30-amino acid segment and the display of these amino acid sequences on an idealized alpha helix revealed a periodic repetition of leucine residues at every seventh position over a distance covering eight helical turns. The polypeptide segments containing these periodic arrays of leucine residues were proposed to exist in an alpha-helical conformation and the leucine side chains from one alpha helix interdigitate with those from the alpha helix of a second polypeptide, facilitating dimerization.
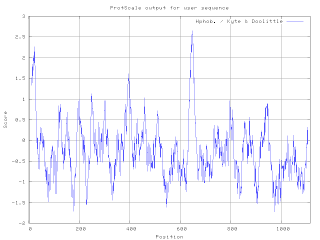
A hydrophilicity plot is a quantitative analysis of the degree of hydrophobicity or hydrophilicity of amino acids of a protein. It is used to characterize or identify possible structure or domains of a protein.

A protein contact map represents the distance between all possible amino acid residue pairs of a three-dimensional protein structure using a binary two-dimensional matrix. For two residues and , the element of the matrix is 1 if the two residues are closer than a predetermined threshold, and 0 otherwise. Various contact definitions have been proposed: The distance between the Cα-Cα atom with threshold 6-12 Å; distance between Cβ-Cβ atoms with threshold 6-12 Å ; and distance between the side-chain centers of mass.
SOSUI is a free online tool that predicts a part of the secondary structure of proteins from a given amino acid sequence (AAS). The main objective is to determine whether the protein in question is a soluble or a transmembrane protein.

A 310 helix is a type of secondary structure found in proteins and polypeptides. Of the numerous protein secondary structures present, the 310-helix is the fourth most common type observed; following α-helices, β-sheets and reverse turns. 310-helices constitute nearly 10–15% of all helices in protein secondary structures, and are typically observed as extensions of α-helices found at either their N- or C- termini. Because of the α-helices tendency to consistently fold and unfold, it has been proposed that the 310-helix serves as an intermediary conformation of sorts, and provides insight into the initiation of α-helix folding.
A helix bundle is a small protein fold composed of several alpha helices that are usually nearly parallel or antiparallel to each other.
The Chou–Fasman method is an empirical technique for the prediction of secondary structures in proteins, originally developed in the 1970s by Peter Y. Chou and Gerald D. Fasman. The method is based on analyses of the relative frequencies of each amino acid in alpha helices, beta sheets, and turns based on known protein structures solved with X-ray crystallography. From these frequencies a set of probability parameters were derived for the appearance of each amino acid in each secondary structure type, and these parameters are used to predict the probability that a given sequence of amino acids would form a helix, a beta strand, or a turn in a protein. The method is at most about 50–60% accurate in identifying correct secondary structures, which is significantly less accurate than the modern machine learning–based techniques.
The GOR method is an information theory-based method for the prediction of secondary structures in proteins. It was developed in the late 1970s shortly after the simpler Chou–Fasman method. Like Chou–Fasman, the GOR method is based on probability parameters derived from empirical studies of known protein tertiary structures solved by X-ray crystallography. However, unlike Chou–Fasman, the GOR method takes into account not only the propensities of individual amino acids to form particular secondary structures, but also the conditional probability of the amino acid to form a secondary structure given that its immediate neighbors have already formed that structure. The method is therefore essentially Bayesian in its analysis.
The Hopp–Woods hydrophilicity scale of amino acids is a method of ranking the amino acids in a protein according to their water solubility in order to search for surface locations on proteins, and especially those locations that tend to form strong interactions with other macromolecules such as proteins, DNA, and RNA.

The chemical shift index or CSI is a widely employed technique in protein nuclear magnetic resonance spectroscopy that can be used to display and identify the location as well as the type of protein secondary structure found in proteins using only backbone chemical shift data The technique was invented by David S. Wishart in 1992 for analyzing 1Hα chemical shifts and then later extended by him in 1994 to incorporate 13C backbone shifts. The original CSI method makes use of the fact that 1Hα chemical shifts of amino acid residues in helices tends to be shifted upfield relative to their random coil values and downfield in beta strands. Similar kinds of upfield and downfield trends are also detectable in backbone 13C chemical shifts.
PSI-blast based secondary structure PREDiction (PSIPRED) is a method used to investigate protein structure. It uses artificial neural network machine learning methods in its algorithm. It is a server-side program, featuring a website serving as a front-end interface, which can predict a protein's secondary structure from the primary sequence.
Membranome database provides structural and functional information about more than 6000 single-pass (bitopic) transmembrane proteins from Homo sapiens, Arabidopsis thaliana, Dictyostelium discoideum, Saccharomyces cerevisiae, Escherichia coli and Methanocaldococcus jannaschii. Bitopic membrane proteins consist of a single transmembrane alpha-helix connecting water-soluble domains of the protein situated at the opposite sides of a biological membrane. These proteins are frequently involved in the signal transduction and communication between cells in multicellular organisms.
The QTY Code is a design method to transform membrane proteins that are intrinsically insoluble in water into variants with water solubility, while retaining their structure and function.
