
Bioinformatics is an interdisciplinary field of science that develops methods and software tools for understanding biological data, especially when the data sets are large and complex. Bioinformatics uses biology, chemistry, physics, computer science, computer programming, information engineering, mathematics and statistics to analyze and interpret biological data. The subsequent process of analyzing and interpreting data is referred to as computational biology.
In molecular biology, restriction fragment length polymorphism (RFLP) is a technique that exploits variations in homologous DNA sequences, known as polymorphisms, populations, or species or to pinpoint the locations of genes within a sequence. The term may refer to a polymorphism itself, as detected through the differing locations of restriction enzyme sites, or to a related laboratory technique by which such differences can be illustrated. In RFLP analysis, a DNA sample is digested into fragments by one or more restriction enzymes, and the resulting restriction fragments are then separated by gel electrophoresis according to their size.
In bioinformatics, sequence assembly refers to aligning and merging fragments from a longer DNA sequence in order to reconstruct the original sequence. This is needed as DNA sequencing technology might not be able to 'read' whole genomes in one go, but rather reads small pieces of between 20 and 30,000 bases, depending on the technology used. Typically, the short fragments (reads) result from shotgun sequencing genomic DNA, or gene transcript (ESTs).
In computational biology, gene prediction or gene finding refers to the process of identifying the regions of genomic DNA that encode genes. This includes protein-coding genes as well as RNA genes, but may also include prediction of other functional elements such as regulatory regions. Gene finding is one of the first and most important steps in understanding the genome of a species once it has been sequenced.
In molecular biology, open reading frames (ORFs) are defined as spans of DNA sequence between the start and stop codons. Usually, this is considered within a studied region of a prokaryotic DNA sequence, where only one of the six possible reading frames will be "open". Such an ORF may contain a start codon and by definition cannot extend beyond a stop codon. That start codon indicates where translation may start. The transcription termination site is located after the ORF, beyond the translation stop codon. If transcription were to cease before the stop codon, an incomplete protein would be made during translation.
User Interface Toolkit (UIT) is a discontinued object-oriented layer that was implemented in C++ programming language atop the XView graphical toolkit. It was developed by Sun Microsystems employees Mark Soloway and Joe Warzecha as an internal tools project for Sun's Computer Integrated Manufacturing organization in 1990.

Multiple sequence alignment (MSA) may refer to the process or the result of sequence alignment of three or more biological sequences, generally protein, DNA, or RNA. In many cases, the input set of query sequences are assumed to have an evolutionary relationship by which they share a linkage and are descended from a common ancestor. From the resulting MSA, sequence homology can be inferred and phylogenetic analysis can be conducted to assess the sequences' shared evolutionary origins. Visual depictions of the alignment as in the image at right illustrate mutation events such as point mutations that appear as differing characters in a single alignment column, and insertion or deletion mutations that appear as hyphens in one or more of the sequences in the alignment. Multiple sequence alignment is often used to assess sequence conservation of protein domains, tertiary and secondary structures, and even individual amino acids or nucleotides.

In bioinformatics a dot plot is a graphical method for comparing two biological sequences and identifying regions of close similarity after sequence alignment. It is a type of recurrence plot.

In bioinformatics, k-mers are substrings of length contained within a biological sequence. Primarily used within the context of computational genomics and sequence analysis, in which k-mers are composed of nucleotides, k-mers are capitalized upon to assemble DNA sequences, improve heterologous gene expression, identify species in metagenomic samples, and create attenuated vaccines. Usually, the term k-mer refers to all of a sequence's subsequences of length , such that the sequence AGAT would have four monomers, three 2-mers, two 3-mers and one 4-mer (AGAT). More generally, a sequence of length will have k-mers and total possible k-mers, where is number of possible monomers.
An operational taxonomic unit (OTU) is an operational definition used to classify groups of closely related individuals. The term was originally introduced in 1963 by Robert R. Sokal and Peter H. A. Sneath in the context of numerical taxonomy, where an "operational taxonomic unit" is simply the group of organisms currently being studied. In this sense, an OTU is a pragmatic definition to group individuals by similarity, equivalent to but not necessarily in line with classical Linnaean taxonomy or modern evolutionary taxonomy.

David Tudor Jones is a Professor of Bioinformatics, and Head of Bioinformatics Group in the University College London. He is also the director in Bloomsbury Center for Bioinformatics, which is a joint Research Centre between UCL and Birkbeck, University of London and which also provides bioinformatics training and support services to biomedical researchers. In 2013, he is a member of editorial boards for PLoS ONE, BioData Mining, Advanced Bioinformatics, Chemical Biology & Drug Design, and Protein: Structure, Function and Bioinformatics.
Community fingerprinting is a set of molecular biology techniques that can be used to quickly profile the diversity of a microbial community. Rather than directly identifying or counting individual cells in an environmental sample, these techniques show how many variants of a gene are present. In general, it is assumed that each different gene variant represents a different type of microbe. Community fingerprinting is used by microbiologists studying a variety of microbial systems to measure biodiversity or track changes in community structure over time. The method analyzes environmental samples by assaying genomic DNA. This approach offers an alternative to microbial culturing, which is important because most microbes cannot be cultured in the laboratory. Community fingerprinting does not result in identification of individual microbe species; instead, it presents an overall picture of a microbial community. These methods are now largely being replaced by high throughput sequencing, such as targeted microbiome analysis and metagenomics.
In metagenomics, binning is the process of grouping reads or contigs and assigning them to individual genome. Binning methods can be based on either compositional features or alignment (similarity), or both.
In bioinformatics, alignment-free sequence analysis approaches to molecular sequence and structure data provide alternatives over alignment-based approaches.
Horizontal or lateral gene transfer is the transmission of portions of genomic DNA between organisms through a process decoupled from vertical inheritance. In the presence of HGT events, different fragments of the genome are the result of different evolutionary histories. This can therefore complicate investigations of the evolutionary relatedness of lineages and species. Also, as HGT can bring into genomes radically different genotypes from distant lineages, or even new genes bearing new functions, it is a major source of phenotypic innovation and a mechanism of niche adaptation. For example, of particular relevance to human health is the lateral transfer of antibiotic resistance and pathogenicity determinants, leading to the emergence of pathogenic lineages.
In bioinformatics, a spaced seed is a pattern of relevant and irrelevant positions in a biosequence and a method of approximate string matching that allows for substitutions. They are a straightforward modification to the earliest heuristic-based alignment efforts that allow for minor differences between the sequences of interest. Spaced seeds have been used in homology search., alignment, assembly, and metagenomics. They are usually represented as a sequence of zeroes and ones, where a one indicates relevance and a zero indicates irrelevance at the given position. Some visual representations use pound signs for relevant and dashes or asterisks for irrelevant positions.
DIMPL is a bioinformatic pipeline that enables the extraction and selection of bacterial GC-rich intergenic regions (IGRs) that are enriched for structured non-coding RNAs (ncRNAs). The method of enriching bacterial IGRs for ncRNA motif discovery was first reported for a study in "Genome-wide discovery of structured noncoding RNAs in bacteria".
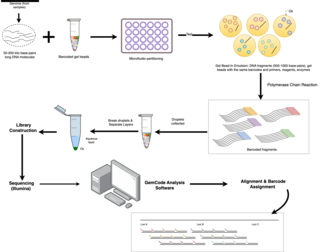
Linked-read sequencing, a type of DNA sequencing technology, uses specialized technique that tags DNA molecules with unique barcodes before fragmenting them. Unlike traditional sequencing technology, where DNA is broken into small fragments and then sequenced individually, resulting in short read lengths that has difficulties in accurately reconstructing the original DNA sequence, the unique barcodes of linked-read sequencing allows scientists to link together DNA fragments that come from the same DNA molecule. A pivotal benefit of this technology lies in the small quantities of DNA required for large genome information output, effectively combining the advantages of long-read and short-read technologies..