Related Research Articles

The striatum, or corpus striatum, is a nucleus in the subcortical basal ganglia of the forebrain. The striatum is a critical component of the motor and reward systems; receives glutamatergic and dopaminergic inputs from different sources; and serves as the primary input to the rest of the basal ganglia.

Huntington's disease (HD), also known as Huntington's chorea, is an incurable neurodegenerative disease that is mostly inherited. The earliest symptoms are often subtle problems with mood or mental/psychiatric abilities. A general lack of coordination and an unsteady gait often follow. It is also a basal ganglia disease causing a hyperkinetic movement disorder known as chorea. As the disease advances, uncoordinated, involuntary body movements of chorea become more apparent. Physical abilities gradually worsen until coordinated movement becomes difficult and the person is unable to talk. Mental abilities generally decline into dementia, depression, apathy, and impulsivity at times. The specific symptoms vary somewhat between people. Symptoms usually begin between 30 and 50 years of age, and can start at any age but are usually seen around the age of 40. The disease may develop earlier in each successive generation. About eight percent of cases start before the age of 20 years, and are known as juvenile HD, which typically present with the slow movement symptoms of Parkinson's disease rather than those of chorea.

Huntingtin(Htt) is the protein coded for in humans by the HTT gene, also known as the IT15 ("interesting transcript 15") gene. Mutated HTT is the cause of Huntington's disease (HD), and has been investigated for this role and also for its involvement in long-term memory storage.

Huntingtin-interacting protein 1 also known as HIP-1 is a protein that in humans is encoded by the HIP1 gene.
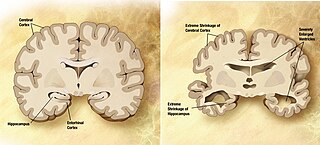
A neurodegenerative disease is caused by the progressive loss of structure or function of neurons, in the process known as neurodegeneration. Such neuronal damage may ultimately involve cell death. Neurodegenerative diseases include amyotrophic lateral sclerosis, multiple sclerosis, Parkinson's disease, Alzheimer's disease, Huntington's disease, multiple system atrophy, tauopathies, and prion diseases. Neurodegeneration can be found in the brain at many different levels of neuronal circuitry, ranging from molecular to systemic. Because there is no known way to reverse the progressive degeneration of neurons, these diseases are considered to be incurable; however research has shown that the two major contributing factors to neurodegeneration are oxidative stress and inflammation. Biomedical research has revealed many similarities between these diseases at the subcellular level, including atypical protein assemblies and induced cell death. These similarities suggest that therapeutic advances against one neurodegenerative disease might ameliorate other diseases as well.

Growth Associated Protein 43 (GAP43) is a protein encoded by the GAP43 gene in humans.

Dynactin subunit 1 is a protein that in humans is encoded by the DCTN1 gene.
Nitric oxide synthase 1 (neuronal), also known as NOS1, is an enzyme that in humans is encoded by the NOS1 gene.

Disks large homolog 2 (DLG2) also known as channel-associated protein of synapse-110 (chapsyn-110) or postsynaptic density protein 93 (PSD-93) is a protein that in humans is encoded by the DLG2 gene.
Kalirin, also known as Huntingtin-associated protein-interacting protein (HAPIP), protein duo (DUO), or serine/threonine-protein kinase with Dbl- and pleckstrin homology domain, is a protein that in humans is encoded by the KALRN gene. Kalirin was first identified in 1997 as a protein interacting with huntingtin-associated protein 1. Is also known to play an important role in nerve growth and axonal development.

Disks large-associated protein 1 (DAP-1), also known as guanylate kinase-associated protein (GKAP), is a protein that in humans is encoded by the DLGAP1 gene. DAP-1 is known to be highly enriched in synaptosomal preparations of the brain, and present in the post-synaptic density.

Nitric oxide synthase 1 adaptor protein (NOS1AP) also known as carboxyl-terminal PDZ ligand of neuronal nitric oxide synthase protein (CAPON) is a protein that in humans is encoded by the NOS1AP gene.
SET domain containing 2 is an enzyme that in humans is encoded by the SETD2 gene.
Intraflagellar transport protein 57 homolog is a protein that in humans is encoded by the IFT57 gene.

Palmitoyltransferase ZDHHC17 is an enzyme that contains a DHHC domain that in humans is encoded by the ZDHHC17 gene.

Kinesin heavy chain isoform 5C is a protein that in humans is encoded by the KIF5C gene. It is part of the kinesin family of motor proteins.

Dentatorubral–pallidoluysian atrophy (DRPLA) is an autosomal dominant spinocerebellar degeneration caused by an expansion of a CAG repeat encoding a polyglutamine tract in the atrophin-1 protein. It is also known as Haw River Syndrome and Naito–Oyanagi disease. Although this condition was perhaps first described by Smith et al. in 1958, and several sporadic cases have been reported from Western countries, this disorder seems to be very rare except in Japan.

Coiled-coil domain-containing protein 113 also known as HSPC065, GC16Pof6842 and GC16P044152, is a protein that in humans is encoded by the CCDC113 gene. The human CCDC113 gene is located on chromosome 16q21 and encodes 5,304 base pairs of mRNA and 377 amino acids.

Basal ganglia disease is a group of physical problems that occur when the group of nuclei in the brain known as the basal ganglia fail to properly suppress unwanted movements or to properly prime upper motor neuron circuits to initiate motor function. Research indicates that increased output of the basal ganglia inhibits thalamocortical projection neurons. Proper activation or deactivation of these neurons is an integral component for proper movement. If something causes too much basal ganglia output, then the ventral anterior (VA) and ventral lateral (VL) thalamocortical projection neurons become too inhibited, and one cannot initiate voluntary movement. These disorders are known as hypokinetic disorders. However, a disorder leading to abnormally low output of the basal ganglia leads to reduced inhibition, and thus excitation, of the thalamocortical projection neurons which synapse onto the cortex. This situation leads to an inability to suppress unwanted movements. These disorders are known as hyperkinetic disorders.
Michelle Gray is an American neuroscientist and assistant professor of neurology and neurobiology at the University of Alabama Birmingham. Gray is a researcher in the study of the biological basis of Huntington's disease (HD). In her postdoctoral work, she developed a transgenic mouse line, BACHD, that is now used worldwide in the study of HD. Gray's research now focuses on the role of glial cells in HD. In 2020 Gray was named one of the 100 Inspiring Black Scientists in America by Cell Press. She is also a member of the Hereditary Disease Foundation’s scientific board.
References
- ↑ Li XJ, Li SH, Sharp AH, Nucifora FC, Schilling G, Lanahan A, Worley P, Snyder SH, Ross CA (November 1995). "A huntingtin-associated protein enriched in brain with implications for pathology". Nature. 378 (6555): 398–402. Bibcode:1995Natur.378..398L. doi:10.1038/378398a0. PMID 7477378. S2CID 4339298.
- ↑ Li SH, Hosseini SH, Gutekunst CA, Hersch SM, Ferrante RJ, Li XJ (July 1998). "A human HAP1 homologue. Cloning, expression, and interaction with huntingtin". J. Biol. Chem. 273 (30): 19220–7. doi: 10.1074/jbc.273.30.19220 . PMID 9668110.
- ↑ "Entrez Gene: HAP1 huntingtin-associated protein 1".
- ↑ Li SH, Li XJ (October 2004). "Huntingtin and its role in neuronal degeneration". Neuroscientist. 10 (5): 467–75. doi:10.1177/1073858404266777. PMID 15359012. S2CID 19491573.
- ↑ Martin EJ, Kim M, Velier J, Sapp E, Lee HS, Laforet G, Won L, Chase K, Bhide PG, Heller A, Aronin N, Difiglia M (January 1999). "Analysis of Huntingtin-associated protein 1 in mouse brain and immortalized striatal neurons". J. Comp. Neurol. 403 (4): 421–30. doi:10.1002/(SICI)1096-9861(19990125)403:4<421::AID-CNE1>3.0.CO;2-5. PMID 9888310. S2CID 20210908.
- ↑ Page KJ, Potter L, Aronni S, Everitt BJ, Dunnett SB (May 1998). "The expression of Huntingtin-associated protein (HAP1) mRNA in developing, adult and ageing rat CNS: implications for Huntington's disease neuropathology". Eur. J. Neurosci. 10 (5): 1835–45. doi:10.1046/j.1460-9568.1998.00185.x. PMID 9751154. S2CID 23547800.
- ↑ Gutekunst CA, Li SH, Yi H, Ferrante RJ, Li XJ, Hersch SM (October 1998). "The cellular and subcellular localization of huntingtin-associated protein 1 (HAP1): comparison with huntingtin in rat and human". J. Neurosci. 18 (19): 7674–86. doi: 10.1523/JNEUROSCI.18-19-07674.1998 . PMC 6793025 . PMID 9742138.
- ↑ Li XJ, Sharp AH, Li SH, Dawson TM, Snyder SH, Ross CA (May 1996). "Huntingtin-associated protein (HAP1): discrete neuronal localizations in the brain resemble those of neuronal nitric oxide synthase". Proc. Natl. Acad. Sci. U.S.A. 93 (10): 4839–44. Bibcode:1996PNAS...93.4839L. doi: 10.1073/pnas.93.10.4839 . PMC 39366 . PMID 8643490.
This article incorporates text from the United States National Library of Medicine, which is in the public domain.