An intron is any nucleotide sequence within a gene that is not expressed or operative in the final RNA product. The word intron is derived from the term intragenic region, i.e., a region inside a gene. The term intron refers to both the DNA sequence within a gene and the corresponding RNA sequence in RNA transcripts. The non-intron sequences that become joined by this RNA processing to form the mature RNA are called exons.
A restriction enzyme, restriction endonuclease, REase, ENase orrestrictase is an enzyme that cleaves DNA into fragments at or near specific recognition sites within molecules known as restriction sites. Restriction enzymes are one class of the broader endonuclease group of enzymes. Restriction enzymes are commonly classified into five types, which differ in their structure and whether they cut their DNA substrate at their recognition site, or if the recognition and cleavage sites are separate from one another. To cut DNA, all restriction enzymes make two incisions, once through each sugar-phosphate backbone of the DNA double helix.

RNA splicing is a process in molecular biology where a newly-made precursor messenger RNA (pre-mRNA) transcript is transformed into a mature messenger RNA (mRNA). It works by removing all the introns and splicing back together exons. For nuclear-encoded genes, splicing occurs in the nucleus either during or immediately after transcription. For those eukaryotic genes that contain introns, splicing is usually needed to create an mRNA molecule that can be translated into protein. For many eukaryotic introns, splicing occurs in a series of reactions which are catalyzed by the spliceosome, a complex of small nuclear ribonucleoproteins (snRNPs). There exist self-splicing introns, that is, ribozymes that can catalyze their own excision from their parent RNA molecule. The process of transcription, splicing and translation is called gene expression, the central dogma of molecular biology.

Ribosomal ribonucleic acid (rRNA) is a type of non-coding RNA which is the primary component of ribosomes, essential to all cells. rRNA is a ribozyme which carries out protein synthesis in ribosomes. Ribosomal RNA is transcribed from ribosomal DNA (rDNA) and then bound to ribosomal proteins to form small and large ribosome subunits. rRNA is the physical and mechanical factor of the ribosome that forces transfer RNA (tRNA) and messenger RNA (mRNA) to process and translate the latter into proteins. Ribosomal RNA is the predominant form of RNA found in most cells; it makes up about 80% of cellular RNA despite never being translated into proteins itself. Ribosomes are composed of approximately 60% rRNA and 40% ribosomal proteins, though this ratio differs between prokaryotes and eukaryotes.

In biology, the word gene has two meanings. The Mendelian gene is a basic unit of heredity. The molecular gene is a sequence of nucleotides in DNA, that is transcribed to produce a functional RNA. There are two types of molecular genes: protein-coding genes and non-coding genes.
In molecular biology, a twintron is an intron-within-intron excised by sequential splicing reactions. A twintron is presumably formed by the insertion of a mobile intron into an existing intron.
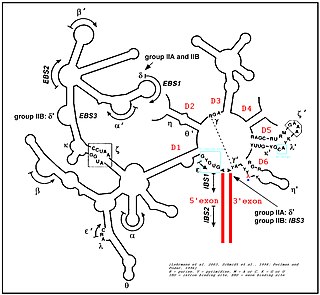
Group II introns are a large class of self-catalytic ribozymes and mobile genetic elements found within the genes of all three domains of life. Ribozyme activity can occur under high-salt conditions in vitro. However, assistance from proteins is required for in vivo splicing. In contrast to group I introns, intron excision occurs in the absence of GTP and involves the formation of a lariat, with an A-residue branchpoint strongly resembling that found in lariats formed during splicing of nuclear pre-mRNA. It is hypothesized that pre-mRNA splicing may have evolved from group II introns, due to the similar catalytic mechanism as well as the structural similarity of the Group II Domain V substructure to the U6/U2 extended snRNA. Finally, their ability to site-specifically insert into DNA sites has been exploited as a tool for biotechnology. For example, group II introns can be modified to make site-specific genome insertions and deliver cargo DNA such as reporter genes or lox sites

The homing endonucleases are a collection of endonucleases encoded either as freestanding genes within introns, as fusions with host proteins, or as self-splicing inteins. They catalyze the hydrolysis of genomic DNA within the cells that synthesize them, but do so at very few, or even singular, locations. Repair of the hydrolyzed DNA by the host cell frequently results in the gene encoding the homing endonuclease having been copied into the cleavage site, hence the term 'homing' to describe the movement of these genes. Homing endonucleases can thereby transmit their genes horizontally within a host population, increasing their allele frequency at greater than Mendelian rates.
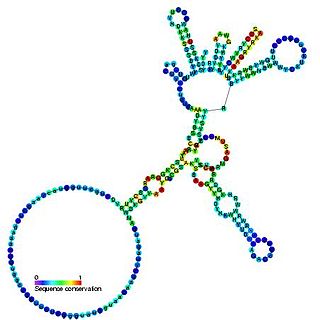
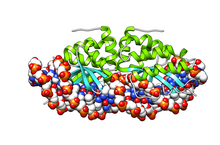
Group I introns are large self-splicing ribozymes. They catalyze their own excision from mRNA, tRNA and rRNA precursors in a wide range of organisms. The core secondary structure consists of nine paired regions (P1-P9). These fold to essentially two domains – the P4-P6 domain and the P3-P9 domain. The secondary structure mark-up for this family represents only this conserved core. Group I introns often have long open reading frames inserted in loop regions.
Artificial gene synthesis, or simply gene synthesis, refers to a group of methods that are used in synthetic biology to construct and assemble genes from nucleotides de novo. Unlike DNA synthesis in living cells, artificial gene synthesis does not require template DNA, allowing virtually any DNA sequence to be synthesized in the laboratory. It comprises two main steps, the first of which is solid-phase DNA synthesis, sometimes known as DNA printing. This produces oligonucleotide fragments that are generally under 200 base pairs. The second step then involves connecting these oligonucleotide fragments using various DNA assembly methods. Because artificial gene synthesis does not require template DNA, it is theoretically possible to make a completely synthetic DNA molecule with no limits on the nucleotide sequence or size.

The 23S rRNA is a 2,904 nucleotide long component of the large subunit (50S) of the bacterial/archean ribosome and makes up the peptidyl transferase center (PTC). The 23S rRNA is divided into six secondary structural domains titled I-VI, with the corresponding 5S rRNA being considered domain VII. The ribosomal peptidyl transferase activity resides in domain V of this rRNA, which is also the most common binding site for antibiotics that inhibit translation, making it a target for ribosomal engineering. A well-known member of this antibiotic class, chloramphenicol, acts by inhibiting peptide bond formation, with recent 3D-structural studies showing two different binding sites depending on the species of ribosome. Numerous mutations in domains of the 23S rRNA with Peptidyl transferase activity have resulted in antibiotic resistance. 23S rRNA genes typically have higher sequence variations, including insertions and/or deletions, compared to other rRNAs.
Ribosomal frameshifting, also known as translational frameshifting or translational recoding, is a biological phenomenon that occurs during translation that results in the production of multiple, unique proteins from a single mRNA. The process can be programmed by the nucleotide sequence of the mRNA and is sometimes affected by the secondary, 3-dimensional mRNA structure. It has been described mainly in viruses, retrotransposons and bacterial insertion elements, and also in some cellular genes.
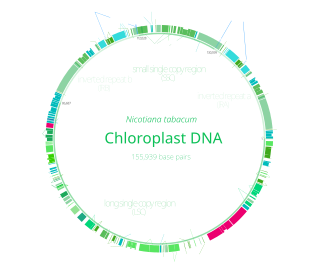
Chloroplast DNA (cpDNA) is the DNA located in chloroplasts, which are photosynthetic organelles located within the cells of some eukaryotic organisms. Chloroplasts, like other types of plastid, contain a genome separate from that in the cell nucleus. The existence of chloroplast DNA was identified biochemically in 1959, and confirmed by electron microscopy in 1962. The discoveries that the chloroplast contains ribosomes and performs protein synthesis revealed that the chloroplast is genetically semi-autonomous. The first complete chloroplast genome sequences were published in 1986, Nicotiana tabacum (tobacco) by Sugiura and colleagues and Marchantia polymorpha (liverwort) by Ozeki et al. Since then, a great number of chloroplast DNAs from various species have been sequenced.
Meganucleases are endodeoxyribonucleases characterized by a large recognition site ; as a result this site generally occurs only once in any given genome. For example, the 18-base pair sequence recognized by the I-SceI meganuclease would on average require a genome twenty times the size of the human genome to be found once by chance. Meganucleases are therefore considered to be the most specific naturally occurring restriction enzymes.
Numerous key discoveries in biology have emerged from studies of RNA, including seminal work in the fields of biochemistry, genetics, microbiology, molecular biology, molecular evolution, and structural biology. As of 2010, 30 scientists have been awarded Nobel Prizes for experimental work that includes studies of RNA. Specific discoveries of high biological significance are discussed in this article.

Genome editing, or genome engineering, or gene editing, is a type of genetic engineering in which DNA is inserted, deleted, modified or replaced in the genome of a living organism. Unlike early genetic engineering techniques that randomly inserts genetic material into a host genome, genome editing targets the insertions to site-specific locations. The basic mechanism involved in genetic manipulations through programmable nucleases is the recognition of target genomic loci and binding of effector DNA-binding domain (DBD), double-strand breaks (DSBs) in target DNA by the restriction endonucleases, and the repair of DSBs through homology-directed recombination (HDR) or non-homologous end joining (NHEJ).