{kind=link}
Related Research Articles
In genetics, shotgun sequencing is a method used for sequencing random DNA strands. It is named by analogy with the rapidly expanding, quasi-random shot grouping of a shotgun.

In molecular biology, a library is a collection of DNA fragments that is stored and propagated in a population of micro-organisms through the process of molecular cloning. There are different types of DNA libraries, including cDNA libraries, genomic libraries and randomized mutant libraries. DNA library technology is a mainstay of current molecular biology, genetic engineering, and protein engineering, and the applications of these libraries depend on the source of the original DNA fragments. There are differences in the cloning vectors and techniques used in library preparation, but in general each DNA fragment is uniquely inserted into a cloning vector and the pool of recombinant DNA molecules is then transferred into a population of bacteria or yeast such that each organism contains on average one construct. As the population of organisms is grown in culture, the DNA molecules contained within them are copied and propagated.
A DNA construct is an artificially-designed segment of DNA borne on a vector that can be used to incorporate genetic material into a target tissue or cell. A DNA construct contains a DNA insert, called a transgene, delivered via a transformation vector which allows the insert sequence to be replicated and/or expressed in the target cell. This gene can be cloned from a naturally occurring gene, or synthetically constructed. The vector can be delivered using physical, chemical or viral methods. Typically, the vectors used in DNA constructs contain an origin of replication, a multiple cloning site, and a selectable marker. Certain vectors can carry additional regulatory elements based on the expression system involved.
A genomic library is a collection of overlapping DNA fragments that together make up the total genomic DNA of a single organism. The DNA is stored in a population of identical vectors, each containing a different insert of DNA. In order to construct a genomic library, the organism's DNA is extracted from cells and then digested with a restriction enzyme to cut the DNA into fragments of a specific size. The fragments are then inserted into the vector using DNA ligase. Next, the vector DNA can be taken up by a host organism - commonly a population of Escherichia coli or yeast - with each cell containing only one vector molecule. Using a host cell to carry the vector allows for easy amplification and retrieval of specific clones from the library for analysis.
A human artificial chromosome (HAC) is a microchromosome that can act as a new chromosome in a population of human cells. That is, instead of 46 chromosomes, the cell could have 47 with the 47th being very small, roughly 6–10 megabases (Mb) in size instead of 50–250 Mb for natural chromosomes, and able to carry new genes introduced by human researchers. Ideally, researchers could integrate different genes that perform a variety of functions, including disease defense.
Fosmids are similar to cosmids but are based on the bacterial F-plasmid. The cloning vector is limited, as a host can only contain one fosmid molecule. Fosmids can hold DNA inserts of up to 40 kb in size; often the source of the insert is random genomic DNA. A fosmid library is prepared by extracting the genomic DNA from the target organism and cloning it into the fosmid vector. The ligation mix is then packaged into phage particles and the DNA is transfected into the bacterial host. Bacterial clones propagate the fosmid library. The low copy number offers higher stability than vectors with relatively higher copy numbers, including cosmids. Fosmids may be useful for constructing stable libraries from complex genomes. Fosmids have high structural stability and have been found to maintain human DNA effectively even after 100 generations of bacterial growth. Fosmid clones were used to help assess the accuracy of the Public Human Genome Sequence.
The Reference Sequence (RefSeq) database is an open access, annotated and curated collection of publicly available nucleotide sequences and their protein products. RefSeq was introduced in 2000. This database is built by National Center for Biotechnology Information (NCBI), and, unlike GenBank, provides only a single record for each natural biological molecule for major organisms ranging from viruses to bacteria to eukaryotes.
Paired-end tags (PET) are the short sequences at the 5’ and 3' ends of a DNA fragment which are unique enough that they (theoretically) exist together only once in a genome, therefore making the sequence of the DNA in between them available upon search or upon further sequencing. Paired-end tags (PET) exist in PET libraries with the intervening DNA absent, that is, a PET "represents" a larger fragment of genomic or cDNA by consisting of a short 5' linker sequence, a short 5' sequence tag, a short 3' sequence tag, and a short 3' linker sequence. It was shown conceptually that 13 base pairs are sufficient to map tags uniquely. However, longer sequences are more practical for mapping reads uniquely. The endonucleases used to produce PETs give longer tags but sequences of 50–100 base pairs would be optimal for both mapping and cost efficiency. After extracting the PETs from many DNA fragments, they are linked (concatenated) together for efficient sequencing. On average, 20–30 tags could be sequenced with the Sanger method, which has a longer read length. Since the tag sequences are short, individual PETs are well suited for next-generation sequencing that has short read lengths and higher throughput. The main advantages of PET sequencing are its reduced cost by sequencing only short fragments, detection of structural variants in the genome, and increased specificity when aligning back to the genome compared to single tags, which involves only one end of the DNA fragment.
Optical mapping is a technique for constructing ordered, genome-wide, high-resolution restriction maps from single, stained molecules of DNA, called "optical maps". By mapping the location of restriction enzyme sites along the unknown DNA of an organism, the spectrum of resulting DNA fragments collectively serves as a unique "fingerprint" or "barcode" for that sequence. Originally developed by Dr. David C. Schwartz and his lab at NYU in the 1990s this method has since been integral to the assembly process of many large-scale sequencing projects for both microbial and eukaryotic genomes. Later technologies use DNA melting, DNA competitive binding or enzymatic labelling in order to create the optical mappings.

DNA barcoding is a method of species identification using a short section of DNA from a specific gene or genes. The premise of DNA barcoding is that by comparison with a reference library of such DNA sections, an individual sequence can be used to uniquely identify an organism to species, just as a supermarket scanner uses the familiar black stripes of the UPC barcode to identify an item in its stock against its reference database. These "barcodes" are sometimes used in an effort to identify unknown species or parts of an organism, simply to catalog as many taxa as possible, or to compare with traditional taxonomy in an effort to determine species boundaries.

Molecular cloning is a set of experimental methods in molecular biology that are used to assemble recombinant DNA molecules and to direct their replication within host organisms. The use of the word cloning refers to the fact that the method involves the replication of one molecule to produce a population of cells with identical DNA molecules. Molecular cloning generally uses DNA sequences from two different organisms: the species that is the source of the DNA to be cloned, and the species that will serve as the living host for replication of the recombinant DNA. Molecular cloning methods are central to many contemporary areas of modern biology and medicine.

Turtle leeches are a genus, Ozobranchus, of leeches (Hirudinea) that feed exclusively on the blood of turtles. Only two species – Ozobranchus margoi and Ozobranchus branchiatus – are found in the Atlantic coast of the United States and the Gulf of Mexico. Little is known about these leeches due to difficulties in studying their sea turtle hosts.

Jumping libraries or junction-fragment libraries are collections of genomic DNA fragments generated by chromosome jumping. These libraries allow the analysis of large areas of the genome and overcome distance limitations in common cloning techniques. A jumping library clone is composed of two stretches of DNA that are usually located many kilobases away from each other. The stretch of DNA located between these two "ends" is deleted by a series of biochemical manipulations carried out at the start of this cloning technique.
The Barcode of Life Data System is a web platform specifically devoted to DNA barcoding. It is a cloud-based data storage and analysis platform developed at the Centre for Biodiversity Genomics in Canada. It consists of four main modules, a data portal, an educational portal, a registry of BINs, and a data collection and analysis workbench which provides an online platform for analyzing DNA sequences. Since its launch in 2005, BOLD has been extended to provide a range of functionality including data organization, validation, visualization and publication. The most recent version of the system, version 4, launched in 2017, brings a set of improvements supporting data collection and analysis but also includes novel functionality improving data dissemination, citation, and annotation. Before November 16, 2020, BOLD already contained barcode sequences for 318,105 formally described species covering animals, plants, fungi, protists.
DNA barcoding is an alternative method to the traditional morphological taxonomic classification, and has frequently been used to identify species of aquatic macroinvertebrates. Many are crucial indicator organisms in the bioassessment of freshwater and marine ecosystems.
DNA barcoding of algae is commonly used for species identification and phylogenetic studies. Algae form a phylogenetically heterogeneous group, meaning that the application of a single universal barcode/marker for species delimitation is unfeasible, thus different markers/barcodes are applied for this aim in different algal groups.
Microbial DNA barcoding is the use of DNA metabarcoding to characterize a mixture of microorganisms. DNA metabarcoding is a method of DNA barcoding that uses universal genetic markers to identify DNA of a mixture of organisms.
DNA barcoding methods for fish are used to identify groups of fish based on DNA sequences within selected regions of a genome. These methods can be used to study fish, as genetic material, in the form of environmental DNA (eDNA) or cells, is freely diffused in the water. This allows researchers to identify which species are present in a body of water by collecting a water sample, extracting DNA from the sample and isolating DNA sequences that are specific for the species of interest. Barcoding methods can also be used for biomonitoring and food safety validation, animal diet assessment, assessment of food webs and species distribution, and for detection of invasive species.
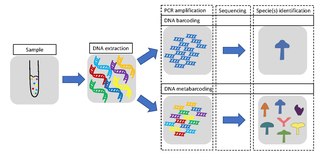
Metabarcoding is the barcoding of DNA/RNA in a manner that allows for the simultaneous identification of many taxa within the same sample. The main difference between barcoding and metabarcoding is that metabarcoding does not focus on one specific organism, but instead aims to determine species composition within a sample.
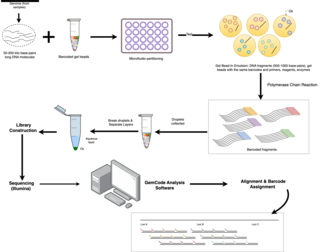
Linked-read sequencing, a type of DNA sequencing technology, uses specialized technique that tags DNA molecules with unique barcodes before fragmenting them. Unlike traditional sequencing technology, where DNA is broken into small fragments and then sequenced individually, resulting in short read lengths that has difficulties in accurately reconstructing the original DNA sequence, the unique barcodes of linked-read sequencing allows scientists to link together DNA fragments that come from the same DNA molecule. A pivotal benefit of this technology lies in the small quantities of DNA required for large genome information output, effectively combining the advantages of long-read and short-read technologies.
References
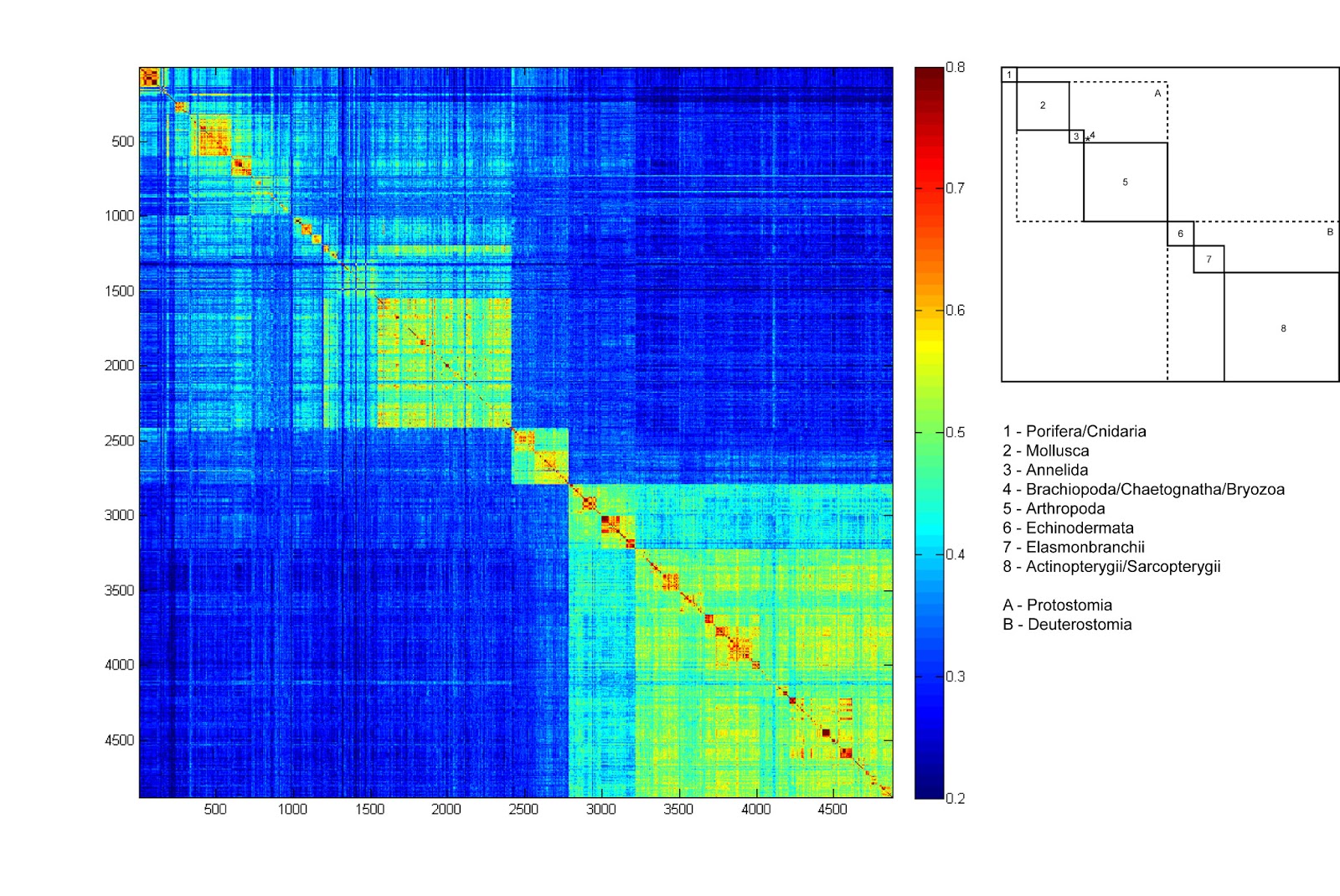
- ↑ "DNA Barcoding: Marine Klee-diagrams (1)". 2013-02-14.