
In atomic physics, the Rutherford–Bohr model or Bohr model or Bohr diagram, presented by Niels Bohr and Ernest Rutherford in 1913, is a system consisting of a small, dense nucleus surrounded by revolving electrons —similar to the structure of the Solar System, but with attraction provided by electrostatic forces rather than gravity. After the cubic model (1902), the plum-pudding model (1904), the Saturnian model (1904), and the Rutherford model (1911) came the Rutherford–Bohr model or just Bohr model for short (1913). The improvement to the Rutherford model is mostly a quantum physical interpretation of it. The model's key success lay in explaining the Rydberg formula for the spectral emission lines of atomic hydrogen. While the Rydberg formula had been known experimentally, it did not gain a theoretical underpinning until the Bohr model was introduced. Not only did the Bohr model explain the reason for the structure of the Rydberg formula, it also provided a justification for its empirical results in terms of fundamental physical constants.
In a chemical reaction, chemical equilibrium is the state in which both reactants and products are present in concentrations which have no further tendency to change with time, so that there is no observable change in the properties of the system. Usually, this state results when the forward reaction proceeds at the same rate as the reverse reaction. The reaction rates of the forward and backward reactions are generally not zero, but equal. Thus, there are no net changes in the concentrations of the reactant(s) and product(s). Such a state is known as dynamic equilibrium.

A covalent bond, also called a molecular bond, is a chemical bond that involves the sharing of electron pairs between atoms. These electron pairs are known as shared pairs or bonding pairs, and the stable balance of attractive and repulsive forces between atoms, when they share electrons, is known as covalent bonding. For many molecules, the sharing of electrons allows each atom to attain the equivalent of a full outer shell, corresponding to a stable electronic configuration.
In quantum chemistry and molecular physics, the Born–Oppenheimer (BO) approximation is the assumption that the motion of atomic nuclei and electrons in a molecule can be separated. The approach is named after Max Born, and J. Robert Oppenheimer. In mathematical terms, it allows the wavefunction of a molecule to be broken into its electronic and nuclear components.
In physics, screening is the damping of electric fields caused by the presence of mobile charge carriers. It is an important part of the behavior of charge-carrying fluids, such as ionized gases, electrolytes, and charge carriers in electronic conductors . In a fluid, with a given permittivity ε, composed of electrically charged constituent particles, each pair of particles interact through the Coulomb force as
Density functional theory (DFT) is a computational quantum mechanical modelling method used in physics, chemistry and materials science to investigate the electronic structure of many-body systems, in particular atoms, molecules, and the condensed phases. Using this theory, the properties of a many-electron system can be determined by using functionals, i.e. functions of another function, which in this case is the spatially dependent electron density. Hence the name density functional theory comes from the use of functionals of the electron density. DFT is among the most popular and versatile methods available in condensed-matter physics, computational physics, and computational chemistry.

The Stark effect is the shifting and splitting of spectral lines of atoms and molecules due to the presence of an external electric field. It is the electric-field analogue of the Zeeman effect, where a spectral line is split into several components due to the presence of the magnetic field. Although initially coined for the static case, it is also used in the wider context to describe effect of time-dependent electric fields. In particular, the Stark effect is responsible for the pressure broadening of spectral lines by charged particles in plasmas. For majority of spectral lines, the Stark effect is either linear or quadratic with a high accuracy.
HSAB concept is an initialism for "hard and soft (Lewis) acids and bases". Also known as the Pearson acid-base concept, HSAB is widely used in chemistry for explaining stability of compounds, reaction mechanisms and pathways. It assigns the terms 'hard' or 'soft', and 'acid' or 'base' to chemical species. 'Hard' applies to species which are small, have high charge states, and are weakly polarizable. 'Soft' applies to species which are big, have low charge states and are strongly polarizable. The concept is a way of applying the notion of orbital overlap to specific chemical cases.
A multipole expansion is a mathematical series representing a function that depends on angles—usually the two angles on a sphere. These series are useful because they can often be truncated, meaning that only the first few terms need to be retained for a good approximation to the original function. The function being expanded may be complex in general. Multipole expansions are very frequently used in the study of electromagnetic and gravitational fields, where the fields at distant points are given in terms of sources in a small region. The multipole expansion with angles is often combined with an expansion in radius. Such a combination gives an expansion describing a function throughout three-dimensional space.
Jellium, also known as the uniform electron gas (UEG) or homogeneous electron gas (HEG), is a quantum mechanical model of interacting electrons in a solid where the positive charges are assumed to be uniformly distributed in space whence the electron density is a uniform quantity as well in space. This model allows one to focus on the effects in solids that occur due to the quantum nature of electrons and their mutual repulsive interactions without explicit introduction of the atomic lattice and structure making up a real material. Jellium is often used in solid-state physics as a simple model of delocalized electrons in a metal, where it can qualitatively reproduce features of real metals such as screening, plasmons, Wigner crystallization and Friedel oscillations.
Mulliken charges arise from the Mulliken population analysis and provide a means of estimating partial atomic charges from calculations carried out by the methods of computational chemistry, particularly those based on the linear combination of atomic orbitals molecular orbital method, and are routinely used as variables in linear regression (QSAR) procedures. The method was developed by Robert S. Mulliken, after whom the method is named. If the coefficients of the basis functions in the molecular orbital are Cμi for the μ'th basis function in the i'th molecular orbital, the density matrix terms are:
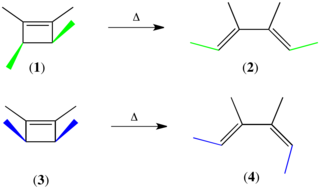
The Woodward–Hoffmann rules, devised by Robert Burns Woodward and Roald Hoffmann, are a set of rules used to rationalize or predict certain aspects of the stereochemistry and activation energy of pericyclic reactions, an important class of reactions in organic chemistry. The Woodward–Hoffmann rules are a consequence of the changes in electronic structure that occur during a pericyclic reaction and are predicated on the phasing of the interacting molecular orbitals. They are applicable to all classes of pericyclic reactions, including (1) electrocyclizations, (2) cycloadditions, (3) sigmatropic reactions, (4) group transfer reactions, (5) ene reactions, (6) cheletropic reactions, and (7) dyotropic reactions. Due to their elegance, simplicity, and generality, the Woodward–Hoffmann rules are credited with first exemplifying the power of molecular orbital theory to experimental chemists.
The Hückel method or Hückel molecular orbital theory, proposed by Erich Hückel in 1930, is a very simple linear combination of atomic orbitals molecular orbitals method for the determination of energies of molecular orbitals of π-electrons in π-delocalized molecules, such as ethylene, benzene, butadiene, and pyridine. It is the theoretical basis for Hückel's rule for the aromaticity of π-electron cyclic, planar systems. It was later extended to conjugated molecules such as pyridine, pyrrole and furan that contain atoms other than carbon, known in this context as heteroatoms. A more dramatic extension of the method to include σ-electrons, known as the extended Hückel method, was developed by Roald Hoffmann. The extended Hückel method gives some degree of quantitative accuracy for organic molecules in general and was used to test the Woodward–Hoffmann rules.
Ewald summation, named after Paul Peter Ewald, is a method for computing long-range interactions in periodic systems. It was first developed as the method for calculating electrostatic energies of ionic crystals, and is now commonly used for calculating long-range interactions in computational chemistry. Ewald summation is a special case of the Poisson summation formula, replacing the summation of interaction energies in real space with an equivalent summation in Fourier space. In this method, the long-range interaction is divided into two parts: a short-range contribution, and a long-range contribution which does not have a singularity. The short-range contribution is calculated in real space, whereas the long-range contribution is calculated using a Fourier transform. The advantage of this method is the rapid convergence of the energy compared with that of a direct summation. This means that the method has high accuracy and reasonable speed when computing long-range interactions, and it is thus the de facto standard method for calculating long-range interactions in periodic systems. The method requires charge neutrality of the molecular system in order to calculate accurately the total Coulombic interaction. A study of the truncation errors introduced in the energy and force calculations of disordered point-charge systems is provided by Kolafa and Perram.
In atomic, molecular, and optical physics and quantum chemistry, the molecular Hamiltonian is the Hamiltonian operator representing the energy of the electrons and nuclei in a molecule. This operator and the associated Schrödinger equation play a central role in computational chemistry and physics for computing properties of molecules and aggregates of molecules, such as thermal conductivity, specific heat, electrical conductivity, optical, and magnetic properties, and reactivity.
The hybrid QM/MM approach is a molecular simulation method that combines the strengths of the QM (accuracy) and MM (speed) approaches, thus allowing for the study of chemical processes in solution and in proteins. The QM/MM approach was introduced in the 1976 paper of Warshel and Levitt. They, along with Martin Karplus, won the 2013 Nobel Prize in Chemistry for "the development of multiscale models for complex chemical systems".
In chemistry, frontier molecular orbital theory is an application of MO theory describing HOMO/LUMO interactions.
In the natural sciences, an intermolecular force is an attraction between two molecules or atoms. They occur from either momentary interactions between molecules or permanent electrostatic attractions between dipoles. They can be explained using a simple phenomenological approach, or using a quantum mechanical approach.
Heat transfer physics describes the kinetics of energy storage, transport, and energy transformation by principal energy carriers: phonons, electrons, fluid particles, and photons. Heat is energy stored in temperature-dependent motion of particles including electrons, atomic nuclei, individual atoms, and molecules. Heat is transferred to and from matter by the principal energy carriers. The state of energy stored within matter, or transported by the carriers, is described by a combination of classical and quantum statistical mechanics. The energy is also transformed (converted) among various carriers. The heat transfer processes are governed by the rates at which various related physical phenomena occur, such as the rate of particle collisions in classical mechanics. These various states and kinetics determine the heat transfer, i.e., the net rate of energy storage or transport. Governing these process from the atomic level to macroscale are the laws of thermodynamics, including conservation of energy.









