Related Research Articles

In organic chemistry, a carboxylic acid is an organic acid that contains a carboxyl group attached to an R-group. The general formula of a carboxylic acid is often written as R−COOH or R−CO2H, sometimes as R−C(O)OH with R referring to an organyl group, or hydrogen, or other groups. Carboxylic acids occur widely. Important examples include the amino acids and fatty acids. Deprotonation of a carboxylic acid gives a carboxylate anion.
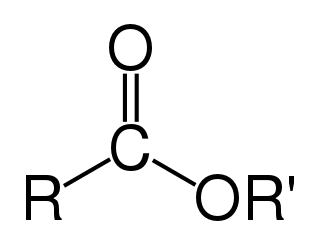
In chemistry, an ester is a functional group derived from an acid in which the hydrogen atom (H) of at least one acidic hydroxyl group of that acid is replaced by an organyl group. Analogues derived from oxygen replaced by other chalcogens belong to the ester category as well. According to some authors, organyl derivatives of acidic hydrogen of other acids are esters as well, but not according to the IUPAC.

In organic chemistry, a ketene is an organic compound of the form RR'C=C=O, where R and R' are two arbitrary monovalent chemical groups. The name may also refer to the specific compound ethenone H2C=C=O, the simplest ketene.

Organometallic chemistry is the study of organometallic compounds, chemical compounds containing at least one chemical bond between a carbon atom of an organic molecule and a metal, including alkali, alkaline earth, and transition metals, and sometimes broadened to include metalloids like boron, silicon, and selenium, as well. Aside from bonds to organyl fragments or molecules, bonds to 'inorganic' carbon, like carbon monoxide, cyanide, or carbide, are generally considered to be organometallic as well. Some related compounds such as transition metal hydrides and metal phosphine complexes are often included in discussions of organometallic compounds, though strictly speaking, they are not necessarily organometallic. The related but distinct term "metalorganic compound" refers to metal-containing compounds lacking direct metal-carbon bonds but which contain organic ligands. Metal β-diketonates, alkoxides, dialkylamides, and metal phosphine complexes are representative members of this class. The field of organometallic chemistry combines aspects of traditional inorganic and organic chemistry.

Formic acid, systematically named methanoic acid, is the simplest carboxylic acid, and has the chemical formula HCOOH and structure H−C(=O)−O−H. It is an important intermediate in chemical synthesis and occurs naturally, most notably in some ants. Esters, salts and the anion derived from formic acid are called formates. Industrially, formic acid is produced from methanol.

Hydrogenation is a chemical reaction between molecular hydrogen (H2) and another compound or element, usually in the presence of a catalyst such as nickel, palladium or platinum. The process is commonly employed to reduce or saturate organic compounds. Hydrogenation typically constitutes the addition of pairs of hydrogen atoms to a molecule, often an alkene. Catalysts are required for the reaction to be usable; non-catalytic hydrogenation takes place only at very high temperatures. Hydrogenation reduces double and triple bonds in hydrocarbons.
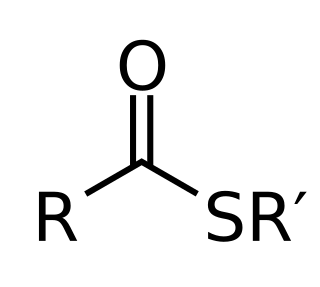
In organic chemistry, thioesters are organosulfur compounds with the molecular structure R−C(=O)−S−R’. They are analogous to carboxylate esters with the sulfur in the thioester replacing oxygen in the carboxylate ester, as implied by the thio- prefix. They are the product of esterification of a carboxylic acid with a thiol. In biochemistry, the best-known thioesters are derivatives of coenzyme A, e.g., acetyl-CoA. The R and R' represent organyl groups, or H in the case of R.
The Friedel–Crafts reactions are a set of reactions developed by Charles Friedel and James Crafts in 1877 to attach substituents to an aromatic ring. Friedel–Crafts reactions are of two main types: alkylation reactions and acylation reactions. Both proceed by electrophilic aromatic substitution.
Alkylation is a chemical reaction that entails transfer of an alkyl group. The alkyl group may be transferred as an alkyl carbocation, a free radical, a carbanion, or a carbene. Alkylating agents are reagents for effecting alkylation. Alkyl groups can also be removed in a process known as dealkylation. Alkylating agents are often classified according to their nucleophilic or electrophilic character. In oil refining contexts, alkylation refers to a particular alkylation of isobutane with olefins. For upgrading of petroleum, alkylation produces a premium blending stock for gasoline. In medicine, alkylation of DNA is used in chemotherapy to damage the DNA of cancer cells. Alkylation is accomplished with the class of drugs called alkylating antineoplastic agents.

Fischer esterification or Fischer–Speier esterification is a special type of esterification by refluxing a carboxylic acid and an alcohol in the presence of an acid catalyst. The reaction was first described by Emil Fischer and Arthur Speier in 1895. Most carboxylic acids are suitable for the reaction, but the alcohol should generally be primary or secondary. Tertiary alcohols are prone to elimination. Contrary to common misconception found in organic chemistry textbooks, phenols can also be esterified to give good to near quantitative yield of products. Commonly used catalysts for a Fischer esterification include sulfuric acid, p-toluenesulfonic acid, and Lewis acids such as scandium(III) triflate. For more valuable or sensitive substrates other, milder procedures such as Steglich esterification are used. The reaction is often carried out without a solvent or in a non-polar solvent that can facilitate Dean–Stark distillation to remove the water byproduct. Typical reaction times vary from 1–10 hours at temperatures of 60–110 °C.
In organic chemistry, hydroformylation, also known as oxo synthesis or oxo process, is an industrial process for the production of aldehydes from alkenes. This chemical reaction entails the net addition of a formyl group and a hydrogen atom to a carbon-carbon double bond. This process has undergone continuous growth since its invention: production capacity reached 6.6×106 tons in 1995. It is important because aldehydes are easily converted into many secondary products. For example, the resultant aldehydes are hydrogenated to alcohols that are converted to detergents. Hydroformylation is also used in speciality chemicals, relevant to the organic synthesis of fragrances and pharmaceuticals. The development of hydroformylation is one of the premier achievements of 20th-century industrial chemistry.

Metal carbonyls are coordination complexes of transition metals with carbon monoxide ligands. Metal carbonyls are useful in organic synthesis and as catalysts or catalyst precursors in homogeneous catalysis, such as hydroformylation and Reppe chemistry. In the Mond process, nickel tetracarbonyl is used to produce pure nickel. In organometallic chemistry, metal carbonyls serve as precursors for the preparation of other organometallic complexes.

The Dakin oxidation (or Dakin reaction) is an organic redox reaction in which an ortho- or para-hydroxylated phenyl aldehyde (2-hydroxybenzaldehyde or 4-hydroxybenzaldehyde) or ketone reacts with hydrogen peroxide (H2O2) in base to form a benzenediol and a carboxylate. Overall, the carbonyl group is oxidised, whereas the H2O2 is reduced.

The Prins reaction is an organic reaction consisting of an electrophilic addition of an aldehyde or ketone to an alkene or alkyne followed by capture of a nucleophile or elimination of an H+ ion. The outcome of the reaction depends on reaction conditions. With water and a protic acid such as sulfuric acid as the reaction medium and formaldehyde the reaction product is a 1,3-diol (3). When water is absent, the cationic intermediate loses a proton to give an allylic alcohol (4). With an excess of formaldehyde and a low reaction temperature the reaction product is a dioxane (5). When water is replaced by acetic acid the corresponding esters are formed.
In chemistry, carbonylation refers to reactions that introduce carbon monoxide (CO) into organic and inorganic substrates. Carbon monoxide is abundantly available and conveniently reactive, so it is widely used as a reactant in industrial chemistry. The term carbonylation also refers to oxidation of protein side chains.
In organometallic chemistry, a migratory insertion is a type of reaction wherein two ligands on a metal complex combine. It is a subset of reactions that very closely resembles the insertion reactions, and both are differentiated by the mechanism that leads to the resulting stereochemistry of the products. However, often the two are used interchangeably because the mechanism is sometimes unknown. Therefore, migratory insertion reactions or insertion reactions, for short, are defined not by the mechanism but by the overall regiochemistry wherein one chemical entity interposes itself into an existing bond of typically a second chemical entity e.g.:

Organocobalt chemistry is the chemistry of organometallic compounds containing a carbon to cobalt chemical bond. Organocobalt compounds are involved in several organic reactions and the important biomolecule vitamin B12 has a cobalt-carbon bond. Many organocobalt compounds exhibit useful catalytic properties, the preeminent example being dicobalt octacarbonyl.
Alcohol oxidation is a collection of oxidation reactions in organic chemistry that convert alcohols to aldehydes, ketones, carboxylic acids, and esters where the carbon carries a higher oxidation state. The reaction mainly applies to primary and secondary alcohols. Secondary alcohols form ketones, while primary alcohols form aldehydes or carboxylic acids.
In chemistry, decarbonylation is a type of organic reaction that involves the loss of carbon monoxide (CO). It is often an undesirable reaction, since it represents a degradation. In the chemistry of metal carbonyls, decarbonylation describes a substitution process, whereby a CO ligand is replaced by another ligand.
An insertion reaction is a chemical reaction where one chemical entity interposes itself into an existing bond of typically a second chemical entity e.g.:
References
- ↑ Molnár, Árpád; Olah, George A.; Surya Prakash, G. K. (2017). "Carbonylation and Carboxylation". Hydrocarbon Chemistry. Wiley. pp. 509–568. doi:10.1002/9781119390541.ch7. ISBN 978-1-119-39051-0.
- 1 2 Weissermel, K., Jargen-Arpe, H. In "Syntheses involving carbon monoxide", Industrial Organic Chemistry; VCH Publishers: New York, NY; pp. 141–145. ( ISBN 978-3527320028)
- ↑ Koch, H.; Haaf, W. (1964). "1-Adamantanecarboxylic Acid". Organic Syntheses. 44: 1. doi:10.15227/orgsyn.044.0001.
- ↑ Barton, Victoria; Ward, Steven A.; Chadwick, James; Hill, Alasdair; o'Neill, Paul M. (2010). "Rationale Design of Biotinylated Antimalarial Endoperoxide Carbon Centered Radical Prodrugs for Applications in Proteomics". Journal of Medicinal Chemistry. 53 (11): 4555–4559. doi:10.1021/jm100201j. PMID 20476788.
- ↑ Becker, Calvin L.; Engstrom, Kenneth M.; Kerdesky, Francis A.; Tolle, John C.; Wagaw, Seble H.; Wang, Weifeng (2008). "A Convergent Process for the Preparation of Adamantane 11-β-HSD-1 Inhibitors". Organic Process Research & Development. 12 (6): 1114–1118. doi:10.1021/op800065q.
- ↑ Kubitschke, Jens; Lange, Horst; Strutz, Heinz (2014). "Carboxylic Acids, Aliphatic". Ullmann's Encyclopedia of Industrial Chemistry. pp. 1–18. doi:10.1002/14356007.a05_235.pub2. ISBN 978-3-527-30673-2.
- ↑ Koch, H.; Haaf, W. Ann. 1958, "618", 251–266.Koch, Herbert; Haaf, Wolfgang (1958). "Über die Synthese verzweigter Carbonsäuren nach der Ameisensäure-Methode". Justus Liebigs Annalen der Chemie. 618: 251–266. doi:10.1002/jlac.19586180127.
- ↑ Li, J. J. In "Koch–Haaf carbonylation"; Name Reactions, 4th ed.; Springer, Berlin, 2009; p. 319. ( doi : 10.1007/978-3-642-01053-8_140)
- 1 2 Qiao, K., Yokoyama, C. Cat. Comm. 2006, 7, 450–453. ( doi : 10.1016/j.catcom.2005.12.009)
- ↑ Souma, Yoshie; Sano, Hiroshi; Iyoda, Jun (1973). "Synthesis of tert-carboxylic acids from olefins and carbon monoxide by coppper(I) carbonyl catalyst". The Journal of Organic Chemistry. 38 (11): 2016–2020. doi:10.1021/jo00951a010.)
- ↑ Xu, Q., Imamura, Y., Fujiwara, M., Souma, Y. J. Org. Chem. , 1997, 62, 1594–1598. ( doi : 10.1021/jo9620122)
- ↑ Xu, Q., Souma, Y. Top. Catal. , 1998, 6, 17. ( doi : 10.1023/A:1019158221240)
- ↑ Tsumori, N., Xu, Q., Souma, Y., Mori, H. J. Mol. Cat. A , 2002, 179, 271–77. ( doi : 10.1016/S1381-1169(01)00396-X)
- ↑ Xu, Q., Inoue, S., Tsumori, N., Mori, H., Kameda, M., Fujiwara, M., Souma, Y. J. Mol. Cat. A , 2001, 170, 147. ( doi : 10.1016/S1381-1169(01)00054-1)
- ↑ Stepanov, A. G., Luzgin, M. V., Romannikov, V. N., Zamaraev, K. I. J. Am. Chem. Soc. , 1995, 117, 3615–16. ( doi : 10.1021/ja00117a032)
- ↑ Coleman, G. H.; Craig, David (1932). "p-Tolualdehyde". Organic Syntheses . 12: 80. doi:10.15227/orgsyn.012.0080 .