Related Research Articles

Beta-lactamases (β-lactamases) are enzymes produced by bacteria that provide multi-resistance to beta-lactam antibiotics such as penicillins, cephalosporins, cephamycins, monobactams and carbapenems (ertapenem), although carbapenems are relatively resistant to beta-lactamase. Beta-lactamase provides antibiotic resistance by breaking the antibiotics' structure. These antibiotics all have a common element in their molecular structure: a four-atom ring known as a beta-lactam (β-lactam) ring. Through hydrolysis, the enzyme lactamase breaks the β-lactam ring open, deactivating the molecule's antibacterial properties.

Molecular genetics is a branch of biology that addresses how differences in the structures or expression of DNA molecules manifests as variation among organisms. Molecular genetics often applies an "investigative approach" to determine the structure and/or function of genes in an organism's genome using genetic screens.

Moraxella catarrhalis is a fastidious, nonmotile, Gram-negative, aerobic, oxidase-positive diplococcus that can cause infections of the respiratory system, middle ear, eye, central nervous system, and joints of humans. It causes the infection of the host cell by sticking to the host cell using trimeric autotransporter adhesins.
Macromolecular docking is the computational modelling of the quaternary structure of complexes formed by two or more interacting biological macromolecules. Protein–protein complexes are the most commonly attempted targets of such modelling, followed by protein–nucleic acid complexes.
In biology, the word gene can have several different meanings. The Mendelian gene is a basic unit of heredity and the molecular gene is a sequence of nucleotides in DNA that is transcribed to produce a functional RNA. There are two types of molecular genes: protein-coding genes and non-coding genes.
The omega loop is a non-regular protein structural motif, consisting of a loop of six or more amino acid residues and any amino acid sequence. The defining characteristic is that residues that make up the beginning and end of the loop are close together in space with no intervening lengths of regular secondary structural motifs. It is named after its shape, which resembles the upper-case Greek letter Omega (Ω).
Gene nomenclature is the scientific naming of genes, the units of heredity in living organisms. It is also closely associated with protein nomenclature, as genes and the proteins they code for usually have similar nomenclature. An international committee published recommendations for genetic symbols and nomenclature in 1957. The need to develop formal guidelines for human gene names and symbols was recognized in the 1960s and full guidelines were issued in 1979. Several other genus-specific research communities have adopted nomenclature standards, as well, and have published them on the relevant model organism websites and in scientific journals, including the Trends in Genetics Genetic Nomenclature Guide. Scientists familiar with a particular gene family may work together to revise the nomenclature for the entire set of genes when new information becomes available. For many genes and their corresponding proteins, an assortment of alternate names is in use across the scientific literature and public biological databases, posing a challenge to effective organization and exchange of biological information. Standardization of nomenclature thus tries to achieve the benefits of vocabulary control and bibliographic control, although adherence is voluntary. The advent of the information age has brought gene ontology, which in some ways is a next step of gene nomenclature, because it aims to unify the representation of gene and gene product attributes across all species.
Jack Y. Yang is an American computer scientist and biophysicist. As of 2011, he is the editor-in-chief of the International Journal of Computational Biology and Drug Design.
TOX high mobility group box family member 3, also known as TOX3, is a human gene.
Robert John Raven is an Australian arachnologist, being the Head of Terrestrial Biodiversity and the Senior Curator (Arachnida) at the Queensland Museum. Dr Raven has described many species of spider in Australia and elsewhere, and is spider bite consultant to the Royal Brisbane Hospital, leading to much work on spider toxins.

In molecular biology short linear motifs (SLiMs), linear motifs or minimotifs are short stretches of protein sequence that mediate protein–protein interaction.

A wide variety of non-coding RNAs have been identified in various species of organisms known to science. However, RNAs have also been identified in "metagenomics" sequences derived from samples of DNA or RNA extracted from the environment, which contain unknown species. Initial work in this area detected homologs of known bacterial RNAs in such metagenome samples. Many of these RNA sequences were distinct from sequences within cultivated bacteria, and provide the potential for additional information on the RNA classes to which they belong.
Minimotif Miner is a program and database designed to identify minimotifs in any protein. Minimotifs are short, contiguous peptide sequences that are known to have a function in at least one protein. Minimotifs are also called sequence motifs or short linear motifs or SLiMs. These are generally restricted to one secondary structure element and are less than 15 amino acids in length.
Computational Resources for Drug Discovery (CRDD) is one of the important silico modules of Open Source for Drug Discovery (OSDD). The CRDD web portal provides computer resources related to drug discovery on a single platform. It provides computational resources for researchers in computer-aided drug design, a discussion forum, and resources to maintain a wiki related to drug discovery, predict inhibitors, and predict the ADME-Tox property of molecules. One of the major objectives of CRDD is to promote open source software in the field of chemoinformatics and pharmacoinformatics.
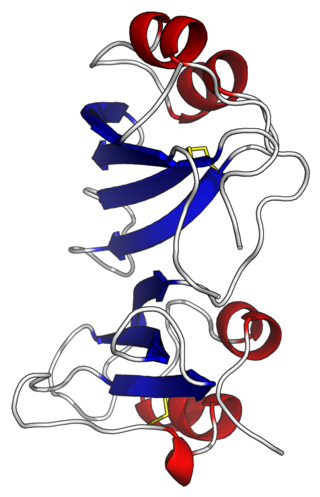
Beta-Lactamase Inhibitor Proteins (BLIPs) are a family of proteins produced by bacterial species including Streptomyces. BLIP acts as a potent inhibitor of beta-lactamases such as TEM-1, which is the most widespread resistance enzyme to penicillin antibiotics. BLIP binds competitively the surface of TEM-1 and inserting residues into the active site to make direct contacts with catalytic residues. BLIP is able to inhibit a variety of class A beta-lactamases, possibly through flexibility of its two domains. The two tandemly repeated domains of BLIP have an α2-β4 structure, the β-hairpin loop from domain 1 inserting into the active site of beta-lactamase. BLIP shows no sequence similarity with BLIP-II, even though both bind to and inhibit TEM-1.

Synaptotagmin XIV is a protein that in humans is encoded by the SYT14 gene.

In evolutionary biology, sequence space is a way of representing all possible sequences. The sequence space has one dimension per amino acid or nucleotide in the sequence leading to highly dimensional spaces.
The Comprehensive Antibiotic Resistance Database (CARD) is a biological database that collects and organizes reference information on antimicrobial resistance genes, proteins and phenotypes. The database covers all types of drug classes and resistance mechanisms and structures its data based on an ontology. The CARD database was one of the first resources that covered antimicrobial resistance genes. The resource is updated monthly and provides tools to allow users to find potential antibiotic resistance genes in newly-sequenced genomes.
The Beta-Lactamase Database (BLAD) is a web-based antimicrobial resistance database that provides structural and phenotypic data on a class of enzymes, beta-lactamase. It hosts sequences from all classes of metallo and non-metallo beta-lactamases. The resource has approximately 2000 gene sequences and compiles its data from various literature, NCBI, protein data bank and other mediums. BLAD is based at the Aligarh Muslim University in the Interdisciplinary Biotechnology Unit. BLAD has four search fields on their site: database, resistance, PDBS, and genome.
CBMAR otherwise known as Comprehensive β-lactamase Molecular Annotation Resource is a database focused on the annotation and discovery of novel beta-lactamase genes and proteins in bacteria. Beta-lactamases are characterized on CBMAR using the Ambler Classification system. CBMAR organizes beta-lactamases according to their classes: A, B, C, and D. They are then further categorized by their (i) sequence variability, (ii) antibiotic resistance profile, (iii) inhibitor susceptibility, (iv) active site, (v) family fingerprints, (vi) mutational profile, (vii) variants, (viii) gene location, (ix) phylogenetic tree, etc. The primary sources of database for CBMAR are GenBank and Uniprot. CBMAR is built on an Apache HTTP Server 2.2.17 with MySQL Ver 14.14 and hosted on Ubuntu 11.04 Linux platform.
References
| | This Biological database-related article is a stub. You can help Wikipedia by expanding it. |