
A cladogram is a diagram used in cladistics to show relations among organisms. A cladogram is not, however, an evolutionary tree because it does not show how ancestors are related to descendants, nor does it show how much they have changed, so many differing evolutionary trees can be consistent with the same cladogram. A cladogram uses lines that branch off in different directions ending at a clade, a group of organisms with a last common ancestor. There are many shapes of cladograms but they all have lines that branch off from other lines. The lines can be traced back to where they branch off. These branching off points represent a hypothetical ancestor which can be inferred to exhibit the traits shared among the terminal taxa above it. This hypothetical ancestor might then provide clues about the order of evolution of various features, adaptation, and other evolutionary narratives about ancestors. Although traditionally such cladograms were generated largely on the basis of morphological characters, DNA and RNA sequencing data and computational phylogenetics are now very commonly used in the generation of cladograms, either on their own or in combination with morphology.

In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns. Sequence alignments are also used for non-biological sequences, such as calculating the distance cost between strings in a natural language or in financial data.
In bioinformatics, neighbor joining is a bottom-up (agglomerative) clustering method for the creation of phylogenetic trees, created by Naruya Saitou and Masatoshi Nei in 1987. Usually based on DNA or protein sequence data, the algorithm requires knowledge of the distance between each pair of taxa to create the phylogenetic tree.
UPGMA is a simple agglomerative (bottom-up) hierarchical clustering method. It also has a weighted variant, WPGMA, and they are generally attributed to Sokal and Michener.
In information theory, linguistics, and computer science, the Levenshtein distance is a string metric for measuring the difference between two sequences. Informally, the Levenshtein distance between two words is the minimum number of single-character edits required to change one word into the other. It is named after the Soviet mathematician Vladimir Levenshtein, who considered this distance in 1965.
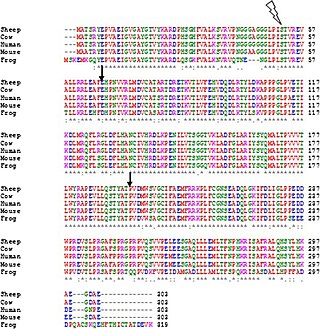
DnaG is a bacterial DNA primase and is encoded by the dnaG gene. The enzyme DnaG, and any other DNA primase, synthesizes short strands of RNA known as oligonucleotides during DNA replication. These oligonucleotides are known as primers because they act as a starting point for DNA synthesis. DnaG catalyzes the synthesis of oligonucleotides that are 10 to 60 nucleotides long, however most of the oligonucleotides synthesized are 11 nucleotides. These RNA oligonucleotides serve as primers, or starting points, for DNA synthesis by bacterial DNA polymerase III. DnaG is important in bacterial DNA replication because DNA polymerase cannot initiate the synthesis of a DNA strand, but can only add nucleotides to a preexisting strand. DnaG synthesizes a single RNA primer at the origin of replication. This primer serves to prime leading strand DNA synthesis. For the other parental strand, the lagging strand, DnaG synthesizes an RNA primer every few kilobases (kb). These primers serve as substrates for the synthesis of Okazaki fragments.

In computer science, a suffix tree is a compressed trie containing all the suffixes of the given text as their keys and positions in the text as their values. Suffix trees allow particularly fast implementations of many important string operations.
In mathematics, computer science and especially graph theory, a distance matrix is a square matrix containing the distances, taken pairwise, between the elements of a set. Depending upon the application involved, the distance being used to define this matrix may or may not be a metric. If there are N elements, this matrix will have size N×N. In graph-theoretic applications the elements are more often referred to as points, nodes or vertices.

In computational linguistics, an n-gram is a contiguous sequence of n items from a given sample of text or speech. The items can be phonemes, syllables, letters, words or base pairs according to the application. The n-grams typically are collected from a text or speech corpus. When the items are words, n-grams may also be called shingles.
FASTA is a DNA and protein sequence alignment software package first described by David J. Lipman and William R. Pearson in 1985. Its legacy is the FASTA format which is now ubiquitous in bioinformatics.
Clustal is a series of widely used computer programs used in bioinformatics for multiple sequence alignment. There have been many versions of Clustal over the development of the algorithm that are listed below. The analysis of each tool and its algorithm is also detailed in their respective categories. Available operating systems listed in the sidebar are a combination of the software availability and may not be supported for every current version of the Clustal tools. Clustal Omega has the widest variety of operating systems out of all the Clustal tools.
In information theory and computer science, the Damerau–Levenshtein distance is a string metric for measuring the edit distance between two sequences. Informally, the Damerau–Levenshtein distance between two words is the minimum number of operations required to change one word into the other.
In computational phylogenetics, tree alignment is a computational problem concerned with producing multiple sequence alignments, or alignments of three or more sequences of DNA, RNA, or protein. Sequences are arranged into a phylogenetic tree, modeling the evolutionary relationships between species or taxa. The edit distances between sequences are calculated for each of the tree's internal vertices, such that the sum of all edit distances within the tree is minimized. Tree alignment can be accomplished using one of several algorithms with various trade-offs between manageable tree size and computational effort.
Intuitively, an algorithmically random sequence is a sequence of binary digits that appears random to any algorithm running on a universal Turing machine. The notion can be applied analogously to sequences on any finite alphabet. Random sequences are key objects of study in algorithmic information theory.
A maximal unique match or MUM, for short, is part of a key step in the multiple sequence alignment of genomes in computational biology. Identification of MUMs and other potential anchors, is the first step in larger alignment systems such as MUMmer. Anchors are the areas between two genomes where they are highly similar. To understand what a MUM is we each word in the acronym can be broken down individually. Match implies that the substring occurs in both sequences to be aligned. Unique means that the substring occurs only once in each sequence. Finally, maximal states that the substring is not part of another larger string that fulfills both prior requirements. The idea behind this, is that long sequences that match exactly and occur only once in each genome are almost certainly part of the global alignment.

In bioinformatics, k-mers are substrings of length contained within a biological sequence. Primarily used within the context of computational genomics and sequence analysis, in which k-mers are composed of nucleotides, k-mers are capitalized upon to assemble DNA sequences, improve heterologous gene expression, identify species in metagenomic samples, and create attenuated vaccines. Usually, the term k-mer refers to all of a sequence's subsequences of length , such that the sequence AGAT would have four monomers, three 2-mers, two 3-mers and one 4-mer (AGAT). More generally, a sequence of length will have k-mers and total possible k-mers, where is number of possible monomers.

Edward Nikolayevich Trifonov is a Russian-born Israeli molecular biophysicist and a founder of Israeli bioinformatics. In his research, he specializes in the recognition of weak signal patterns in biological sequences and is known for his unorthodox scientific methods.
In the field of computational biology, a planted motif search (PMS) also known as a (l, d)-motif search (LDMS) is a method for identifying conserved motifs within a set of nucleic acid or peptide sequences.
In bioinformatics, alignment-free sequence analysis approaches to molecular sequence and structure data provide alternatives over alignment-based approaches.
The Lempel–Ziv complexity is a measure that was first presented in the article On the Complexity of Finite Sequences, by two Israeli computer scientists, Abraham Lempel and Jacob Ziv. This complexity measure is related to Kolmogorov complexity, but the only function it uses is the recursive copy.
