Related Research Articles
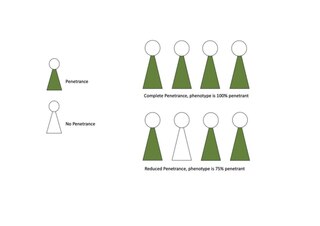
Penetrance in genetics is the proportion of individuals carrying a particular variant of a gene (genotype) that also expresses an associated trait (phenotype). In medical genetics, the penetrance of a disease-causing mutation is the proportion of individuals with the mutation that exhibit clinical symptoms among all individuals with such mutation. For example: If a mutation in the gene responsible for a particular autosomal dominant disorder has 75% penetrance, then 75% of those with the mutation will go on to develop the disease, showing its phenotype, whereas 25% will not.

Retinitis pigmentosa (RP) is a genetic disorder of the eyes that causes loss of vision. Symptoms include trouble seeing at night and decreasing peripheral vision. As peripheral vision worsens, people may experience "tunnel vision". Complete blindness is uncommon. Onset of symptoms is generally gradual and often begins in childhood.

Osteogenesis imperfecta, colloquially known as brittle bone disease, is a group of genetic disorders that all result in bones that break easily. The range of symptoms—on the skeleton as well as on the body's other organs—may be mild to severe. Symptoms found in various types of OI include whites of the eye (sclerae) that are blue instead, short stature, loose joints, hearing loss, breathing problems and problems with the teeth. Potentially life-threatening complications, all of which become more common in more severe OI, include: tearing (dissection) of the major arteries, such as the aorta; pulmonary valve insufficiency secondary to distortion of the ribcage; and basilar invagination.

Haploinsufficiency in genetics describes a model of dominant gene action in diploid organisms, in which a single copy of the wild-type allele at a locus in heterozygous combination with a variant allele is insufficient to produce the wild-type phenotype. Haploinsufficiency may arise from a de novo or inherited loss-of-function mutation in the variant allele, such that it yields little or no gene product. Although the other, standard allele still produces the standard amount of product, the total product is insufficient to produce the standard phenotype. This heterozygous genotype may result in a non- or sub-standard, deleterious, and (or) disease phenotype. Haploinsufficiency is the standard explanation for dominant deleterious alleles.

ATP-binding cassette, sub-family A (ABC1), member 4, also known as ABCA4 or ABCR, is a protein which in humans is encoded by the ABCA4 gene.
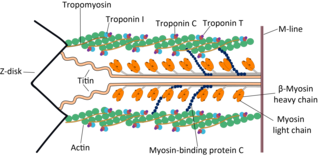
The myosin-binding protein C, cardiac-type is a protein that in humans is encoded by the MYBPC3 gene. This isoform is expressed exclusively in heart muscle during human and mouse development, and is distinct from those expressed in slow skeletal muscle (MYBPC1) and fast skeletal muscle (MYBPC2).

Peripherin-2 is a protein, that in humans is encoded by the PRPH2 gene. Peripherin-2 is found in the rod and cone cells of the retina of the eye. Defects in this protein result in one form of retinitis pigmentosa, an incurable blindness.

PRP31 pre-mRNA processing factor 31 homolog , also known as PRPF31, is a protein which in humans is encoded by the PRPF31 gene.
Nephrocystin-1 is a protein that in humans is encoded by the NPHP1 gene.

Protein XRP2 is a protein that in humans is encoded by the RP2 gene.
Crumbs homolog 1 is a protein that in humans is encoded by the CRB1 gene.

Myosin heavy chain, α isoform (MHC-α) is a protein that in humans is encoded by the MYH6 gene. This isoform is distinct from the ventricular/slow myosin heavy chain isoform, MYH7, referred to as MHC-β. MHC-α isoform is expressed predominantly in human cardiac atria, exhibiting only minor expression in human cardiac ventricles. It is the major protein comprising the cardiac muscle thick filament, and functions in cardiac muscle contraction. Mutations in MYH6 have been associated with late-onset hypertrophic cardiomyopathy, atrial septal defects and sick sinus syndrome.

Rod outer segment membrane protein 1 is a protein that in humans is encoded by the ROM1 gene.

Oxygen-regulated protein 1 also known as retinitis pigmentosa 1 protein (RP1) is a protein that in humans is encoded by the RP1 gene.

NADH dehydrogenase [ubiquinone] flavoprotein 2, mitochondrial (NDUFV2) is an enzyme that in humans is encoded by the NDUFV2 gene. The encoded protein, NDUFV2, is a subunit of complex I of the mitochondrial respiratory chain, which is located on the inner mitochondrial membrane and involved in oxidative phosphorylation. Mutations in this gene are implicated in Parkinson's disease, bipolar disorder, schizophrenia, and have been found in one case of early onset hypertrophic cardiomyopathy and encephalopathy.
Germline mosaicism, also called gonadal mosaicism, is a type of genetic mosaicism where more than one set of genetic information is found specifically within the gamete cells; conversely, somatic mosaicism is a type of genetic mosaicism found in somatic cells. Germline mosaicism can be present at the same time as somatic mosaicism or individually, depending on when the conditions occur. Pure germline mosaicism refers to mosaicism found exclusively in the gametes and not in any somatic cells. Germline mosaicism can be caused either by a mutation that occurs after conception, or by epigenetic regulation, alterations to DNA such as methylation that do not involve changes in the DNA coding sequence.
Genetic heterogeneity occurs through the production of single or similar phenotypes through different genetic mechanisms. There are two types of genetic heterogeneity: allelic heterogeneity, which occurs when a similar phenotype is produced by different alleles within the same gene; and locus heterogeneity, which occurs when a similar phenotype is produced by mutations at different loci.
Mitochondrially encoded tRNA glycine also known as MT-TG is a transfer RNA which in humans is encoded by the mitochondrial MT-TG gene.

Retinal degeneration is a retinopathy which consists in the deterioration of the retina caused by the progressive death of its cells. There are several reasons for retinal degeneration, including artery or vein occlusion, diabetic retinopathy, R.L.F./R.O.P., or disease. These may present in many different ways such as impaired vision, night blindness, retinal detachment, light sensitivity, tunnel vision, and loss of peripheral vision to total loss of vision. Of the retinal degenerative diseases retinitis pigmentosa (RP) is a very important example.
A human disease modifier gene is a modifier gene that alters expression of a human gene at another locus that in turn causes a genetic disease. Whereas medical genetics has tended to distinguish between monogenic traits, governed by simple, Mendelian inheritance, and quantitative traits, with cumulative, multifactorial causes, increasing evidence suggests that human diseases exist on a continuous spectrum between the two.
References
- 1 2 3 4 Keith BP, Robertson DL, Hentges KE (2014-12-09). "Locus heterogeneity disease genes encode proteins with high interconnectivity in the human protein interaction network". Frontiers in Genetics. 5: 434. doi: 10.3389/fgene.2014.00434 . PMC 4260505 . PMID 25538735.
- ↑ Ackermann M (August 2015). "A functional perspective on phenotypic heterogeneity in microorganisms". Nature Reviews. Microbiology. 13 (8): 497–508. doi:10.1038/nrmicro3491. PMID 26145732. S2CID 29846214.
- ↑ "NCI Dictionary of Genetics Terms". National Cancer Institute. 2012-07-20. Retrieved 2019-11-27.
- ↑ Daiger SP, Sullivan LS, Bowne SJ (August 2013). "Genes and mutations causing retinitis pigmentosa". Clinical Genetics. 84 (2): 132–41. doi:10.1111/cge.12203. PMC 3856531 . PMID 23701314.
- ↑ Solomon SD, Jarcho JA, McKenna W, Geisterfer-Lowrance A, Germain R, Salerni R, et al. (September 1990). "Familial hypertrophic cardiomyopathy is a genetically heterogeneous disease". The Journal of Clinical Investigation. 86 (3): 993–9. doi:10.1172/JCI114802. PMC 296820 . PMID 1975599.
- ↑ Van Dijk FS, Sillence DO (June 2014). "Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment". American Journal of Medical Genetics. Part A. 164A (6): 1470–81. doi:10.1002/ajmg.a.36545. PMC 4314691 . PMID 24715559.
- ↑ Goldstein JL, Dana SE, Brunschede GY, Brown MS (March 1975). "Genetic heterogeneity in familial hypercholesterolemia: evidence for two different mutations affecting functions of low-density lipoprotein receptor". Proceedings of the National Academy of Sciences of the United States of America. 72 (3): 1092–6. Bibcode:1975PNAS...72.1092G. doi: 10.1073/pnas.72.3.1092 . PMC 432472 . PMID 236556.
- ↑ Keats, Bronya J. B.; Berlin, Charles I. (1999-01-01). "Genomics and Hearing Impairment". Genome Research. 9 (1): 7–16. doi: 10.1101/gr.9.1.7 . ISSN 1088-9051. PMID 9927480.
- 1 2 3 Rehman AU, Santos-Cortez RL, Drummond MC, Shahzad M, Lee K, Morell RJ, et al. (September 2015). "Challenges and solutions for gene identification in the presence of familial locus heterogeneity". European Journal of Human Genetics. 23 (9): 1207–15. doi:10.1038/ejhg.2014.266. PMC 4538203 . PMID 25491636.
- ↑ Wang D, Huang J (May 2006). "Detecting linkage disequilibrium in the presence of locus heterogeneity". Annals of Human Genetics. 70 (Pt 3): 397–409. doi:10.1111/j.1529-8817.2005.00229.x. PMID 16674561. S2CID 25856694.
- ↑ Pal DK, Greenberg DA (2002). "Evaluating genetic heterogeneity in complex disorders". Human Heredity. 53 (4): 216–26. doi:10.1159/000066195. PMID 12435885. S2CID 46285173.
- ↑ Hartong DT, Berson EL, Dryja TP (November 2006). "Retinitis pigmentosa". Lancet. 368 (9549): 1795–809. doi:10.1016/S0140-6736(06)69740-7. PMID 17113430. S2CID 24950783.
- ↑ "RetNet: Summaries". sph.uth.edu. Retrieved 2019-11-27.
- ↑ Ferrari S, Di Iorio E, Barbaro V, Ponzin D, Sorrentino FS, Parmeggiani F (June 2011). "Retinitis pigmentosa: genes and disease mechanisms". Current Genomics. 12 (4): 238–49. doi:10.2174/138920211795860107. PMC 3131731 . PMID 22131869.
| | This genetics article is a stub. You can help Wikipedia by expanding it. |