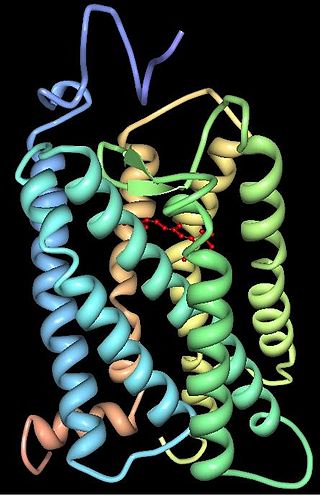
Rhodopsin, also known as visual purple, is a protein encoded by the RHO gene and a G-protein-coupled receptor (GPCR). It is the opsin of the rod cells in the retina and a light-sensitive receptor protein that triggers visual phototransduction in rods. Rhodopsin mediates dim light vision and thus is extremely sensitive to light. When rhodopsin is exposed to light, it immediately photobleaches. In humans, it is regenerated fully in about 30 minutes, after which the rods are more sensitive. Defects in the rhodopsin gene cause eye diseases such as retinitis pigmentosa and congenital stationary night blindness.

Retinitis pigmentosa (RP) is a genetic disorder of the eyes that causes loss of vision. Symptoms include trouble seeing at night and decreasing peripheral vision. As peripheral vision worsens, people may experience "tunnel vision". Complete blindness is uncommon. Onset of symptoms is generally gradual and often begins in childhood.

The photoreceptor cell-specific nuclear receptor (PNR), also known as NR2E3, is a protein that in humans is encoded by the NR2E3 gene. PNR is a member of the nuclear receptor super family of intracellular transcription factors.

RPE-retinal G protein-coupled receptor also known as RGR-opsin is a protein that in humans is encoded by the RGR gene. RGR-opsin is a member of the rhodopsin-like receptor subfamily of GPCR. Like other opsins which bind retinaldehyde, it contains a conserved lysine residue in the seventh transmembrane domain. RGR-opsin comes in different isoforms produced by alternative splicing.

Peripherin-2 is a protein, that in humans is encoded by the PRPH2 gene. Peripherin-2 is found in the rod and cone cells of the retina of the eye. Defects in this protein result in one form of retinitis pigmentosa, an incurable blindness.

Cone-rod homeobox protein is a protein that in humans is encoded by the CRX gene.

Retinaldehyde-binding protein 1 (RLBP1) also known as cellular retinaldehyde-binding protein (CRALBP) is a 36-kD water-soluble protein that in humans is encoded by the RLBP1 gene.

Protein XRP2 is a protein that in humans is encoded by the RP2 gene.

Rod cGMP-specific 3',5'-cyclic phosphodiesterase subunit beta is the beta subunit of the protein complex PDE6 that is encoded by the PDE6B gene. PDE6 is crucial in transmission and amplification of visual signal. The existence of this beta subunit is essential for normal PDE6 functioning. Mutations in this subunit are responsible for retinal degeneration such as retinitis pigmentosa or congenital stationary night blindness.

Neural retina-specific leucine zipper protein is a protein that in humans is encoded by the NRL gene.

Centrosomal protein of 290 kDa is a protein that in humans is encoded by the CEP290 gene. CEP290 is located on the Q arm of chromosome 12.
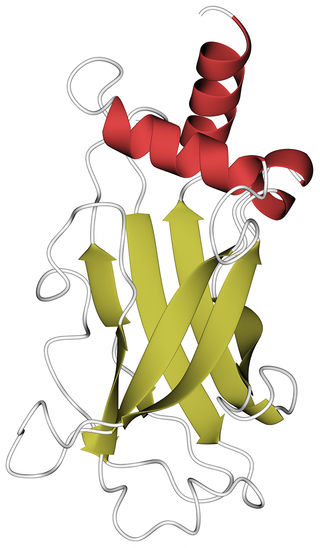
X-linked retinitis pigmentosa GTPase regulator-interacting protein 1 is a protein in the ciliary transition zone that in humans is encoded by the RPGRIP1 gene. RPGRIP1 is a multi-domain protein containing a coiled-coil domain at the N-terminus, two C2 domains and a C-terminal RPGR-interacting domain (RID). Defects in the gene result in the Leber congenital amaurosis (LCA) syndrome and in the eye disease glaucoma.

Rod outer segment membrane protein 1 is a protein that in humans is encoded by the ROM1 gene.

Oxygen-regulated protein 1 also known as retinitis pigmentosa 1 protein (RP1) is a protein that in humans is encoded by the RP1 gene.
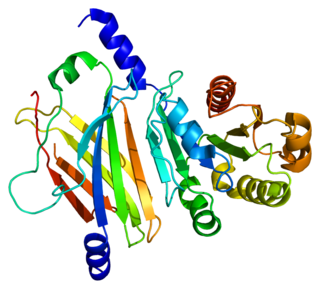
Retinal rod rhodopsin-sensitive cGMP 3',5'-cyclic phosphodiesterase subunit delta is an enzyme that in humans is encoded by the PDE6D gene. PDE6D was originally identified as a fourth subunit of rod cell-specific cGMP phosphodiesterase (PDE). The precise function of PDE delta subunit in the rod specific GMP-PDE complex is unclear. In addition, PDE delta subunit is not confined to photoreceptor cells but is widely distributed in different tissues. PDE delta subunit is thought to be a specific soluble transport factor for certain prenylated proteins and Arl2-GTP a regulator of PDE-mediated transport.
Conorenal syndrome, is a collection of medical conditions that seem to have a common genetic cause.
RPGRIP1L is a human gene.

Retinal degeneration is a retinopathy which consists in the deterioration of the retina caused by the progressive death of its cells. There are several reasons for retinal degeneration, including artery or vein occlusion, diabetic retinopathy, R.L.F./R.O.P., or disease. These may present in many different ways such as impaired vision, night blindness, retinal detachment, light sensitivity, tunnel vision, and loss of peripheral vision to total loss of vision. Of the retinal degenerative diseases retinitis pigmentosa (RP) is a very important example.
Retinal gene therapy holds a promise in treating different forms of non-inherited and inherited blindness.

Pleasantine Mill is a cell biologist and group leader at the MRC Human Genetics Unit at the University of Edinburgh. She won the 2018 British Society for Cell Biology Women in Cell Biology Early Career Medal.




