The cilium is a short hair-like membrane protrusion from many types of eukaryotic cell. The cilium has the shape of a slender threadlike projection that extends from the surface of the much larger cell body. Eukaryotic flagella found on sperm cells and many protozoans have a similar structure to motile cilia that enables swimming through liquids; they are longer than cilia and have a different undulating motion.

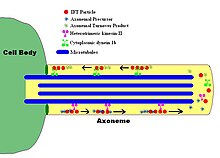
In molecular biology, an axoneme, also called an axial filament, is the microtubule-based cytoskeletal structure that forms the core of a cilium or flagellum. Cilia and flagella are found on many cells, organisms, and microorganisms, to provide motility. The axoneme serves as the "skeleton" of these organelles, both giving support to the structure and, in some cases, the ability to bend. Though distinctions of function and length may be made between cilia and flagella, the internal structure of the axoneme is common to both.

Bardet–Biedl syndrome (BBS) is a ciliopathic human genetic disorder that produces many effects and affects many body systems. It is characterized by rod/cone dystrophy, polydactyly, central obesity, hypogonadism, and kidney dysfunction in some cases. Historically, slower mental processing has also been considered a principal symptom but is now not regarded as such.

Kinesin-like protein KIF3A is a protein that in humans is encoded by the KIF3A gene.

Kinesin-like protein KIF3B is a protein that in humans is encoded by the KIF3B gene. KIF3B is an N-type protein that complexes with two other kinesin proteins to form two-headed anterograde motors. First, KIF3B forms a heterodimer with KIF3A ; (KIF3A/3B), that is membrane-bound and has ATPase activity. Then KIFAP3 binds to the tail domain to form a heterotrimeric motor. This motor has a plus end-directed microtubule sliding activity that exhibits a velocity of ~0.3 μm/s a. There are 14 kinesin protein families in the kinesin superfamily and KIF3B is part of the Kinesin-2 family, of kinesins that can all form heterotrimeric complexes. Expression of the three motor subunits is ubiquitous. The KIG3A/3B/KAP3 motors can transport 90 to 160 nm in diameter organelles.

Intraflagellar transport protein 88 homolog is a protein that is encoded by the IFT88 gene.

Dynein axonemal intermediate chain 1 is a protein that in humans is encoded by the DNAI1 gene.

Intraflagellar transport protein 57 homolog is a protein that in humans is encoded by the IFT57 gene.

Kinesin-like protein KIF17 is a protein that in humans is encoded by the KIF17 gene. KIF17 and its close relative, C. elegans OSM-3, are members of the kinesin-2 family of plus-end directed microtubule-based motor proteins. In contrast to heterotrimeric kinesin-2 motors, however, KIF17 and OSM-3 form distinct homodimeric complexes. Homodimeric kinesin-2 has been implicated in the transport of NMDA receptors along dendrites for delivery to the dendritic membrane, whereas both heterotrimeric and homodimeric kinesin-2 motors function cooperatively in anterograde intraflagellar transport (IFT) and cilium biogenesis.

Intraflagellar transport protein 20 homolog is a protein that in humans is encoded by the IFT20 gene. The gene is composed of 6 exons and is located on human chromosome 17p11.1. This gene is expressed in human brain, lung, kidney and pancreas, and lower expression were also detected in human placenta, liver, thymus, prostate and testis.

A ciliopathy is any genetic disorder that affects the cellular cilia or the cilia anchoring structures, the basal bodies, or ciliary function. Primary cilia are important in guiding the process of development, so abnormal ciliary function while an embryo is developing can lead to a set of malformations that can occur regardless of the particular genetic problem. The similarity of the clinical features of these developmental disorders means that they form a recognizable cluster of syndromes, loosely attributed to abnormal ciliary function and hence called ciliopathies. Regardless of the actual genetic cause, it is clustering of a set of characteristic physiological features which define whether a syndrome is a ciliopathy.

Intraflagellar transport protein 81 homolog is a protein that in humans is encoded by the IFT81 gene. Together with IFT74/72 it forms a core complex to build IFT particles which are required for cilium formation. Additionally, it interacts with basal body components as CEP170 which regulates the disassembly of the cilium.
ADP-ribosylation factor-like protein 13B (ARL13B), also known as ADP-ribosylation factor-like protein 2-like 1, is a protein that in humans is encoded by the ARL13B gene.
A BBSome is a protein complex that operates in primary cilia biogenesis, homeostasis, and intraflagellar transport (IFT). The BBSome recognizes cargo proteins and signaling molecules like G-protein coupled receptors (GPCRs) on the ciliary membrane and helps transport them to and from the primary cilia. Primary cilia are nonmotile microtubule projections that function like antennae and are found in many types of cells. They receive various environmental signals to aid the cell in survival. They can detect photons by concentrating rhodopsin, a light receptor that converts photons into chemical signals, or odorants by concentrating olfactory receptors on the primary cilia surface. Primary cilia are also meaningful in cell development and signaling. They do not contain any way to make proteins within the primary cilia, so the BBSome aids in transporting essential proteins to, from, and within the cilia. Examples of cargo proteins that the BBSome is responsible for ferrying include smoothened, polycystic-1 (PC1), and several G-Protein coupled receptors (GPCRs) like somatostatin receptors (Sstr3), melanin-concentrating hormone receptor 1 (Mchr1), and neuropeptide Y2 receptor.

Sensenbrenner syndrome is a rare multisystem disease first described by Judith A. Sensenbrenner in 1975. It is inherited in an autosomal recessive fashion, and a number of genes appear to be responsible. Three genes responsible have been identified: intraflagellar transport (IFT)122 (WDR10), IFT43—a subunit of the IFT complex A machinery of primary cilia, and WDR35

Ciliogenesis is defined as the building of the cell's antenna or extracellular fluid mediation mechanism. It includes the assembly and disassembly of the cilia during the cell cycle. Cilia are important appendages of cells and are involved in numerous activities such as cell signaling, processing developmental signals, and directing the flow of fluids such as mucus over and around cells. Due to the importance of these cell processes, defects in ciliogenesis can lead to numerous human diseases related to non-functioning cilia known as ciliopathies.

IFT140, Intraflagellar transport 140 homolog, is a protein that in humans is encoded by the IFT140 gene. The gene product forms a core component of IFT-A complex which is indipensible for retrograde intraflagellar transport within the primary cilium.

Tetratricopeptide repeat domain 21B is a protein that in humans is encoded by the TTC21B gene.
RVxP motif is a protein motif involved in localizing proteins into cilia.
Gaia Pigino is the Associate Head of the Structural Biology Research Center and Leader of the "Pigino Group" at the Human Technopole in Milan, Italy.